Vzájemné srovnání statinů z pohledu farmakologa
V současné době je v České republice dostupných šest statinů. Nejčastěji užívanými jsou atorvastatin, simvastatin a rosuvastatin, jejich podíl na celkové spotřebě se pohybuje kolem 95 %. Hlavní rozdíl mezi statiny spočívá ve farmakokinetických vlastnostech. Ty ovlivňují variabilitu jejich hladiny, což se do velké míry odráží v jejich snášenlivosti i délce působení, která zase určuje jejich účinnost.
Lipofilní statiny (atorvastatin, lovastatin a simvastatin) jsou substráty eliminačního transportéru glykoproteinu P (P-gp) a izoenzymu CYP3A4/5 a inhibice či indukce obou systémů významně ovlivní jejich hladinu a snášenlivost. Fluvastatin je substrátem izoenzymu CYP2C9, tato oxidáza je podstatně méně citlivá k ovlivnění aktivity. Hydrofilní statiny (rosuvastatin, pitavastatin a pravastatin) nejsou transformovány izoenzymy CYP a jejich biologická dostupnost není významně ovlivněna P-gp. Variabilita hladiny této skupiny statinů je významně menší.
Vstup všech statinů do místa jejich působení – do hepatocytu – je ovlivněn polymorfní influxní pumpou OATP1B1, transportérem s významnou variabilitou účinnosti. Dostatečný influx statinů do hepatocytu je podmínkou farmakodynamického účinku, tedy inhibice HMG-CoA reduktázy i jejich eliminace do žluče. Nízká aktivita OATP1B1 může vést k nedostatečné učinnosti, a naopak k navození nežádoucích účinků. Zejména u simvastatinu, který má silnou afinitu k této pumpě, je výskyt myalgií a myopatií významně ovlivněn aktivitou transportéru. Při jeho nulové aktivitě stoupá toxicita (zejména výskyt myalgií/myopatií) 20–30násobně.
Statiny jsou kompetitivními inhibitory HMG-CoA reduktázy, jejich dostatečná koncentrace v průběhu dávkovacího období rozhoduje, spolu s dávkou statinu, o výsledném hypolipidemickém účinku. U déle působících statinů (rosuvastatin a atorvastatin) je inhibice aktivity HMG-CoA reduktázy stabilnější a snížení koncentrace LDL cholesterolu výraznější.
Ve farmakoterapii se tyto vlastnosti projeví v míře inhibice syntézy cholesterolu, u dlouhodobě působících statinů (rosuvastatinu a atorvastatinu) je tak efekt na snížení hladiny LDL cholesterolu největší. Variabilita expozice simvastatinu se na straně druhé projeví vyšším výskytem nežádoucích účinků, zejména myalgiemi. Naopak fluvastatin, jehož biologická dostupnost je relativně stabilní a rovněž vstup do hepatocytu je málo závislý na transportéru OATP1B1, je relativně nejlépe snášen. Bohužel z krátkodobého efektu vyplývá i nižší hypolipidemický účinek, v době snížené koncentrace statinu dochází k akceleraci syntézy cholesterolu.
Statiny jsou jednou z nejčastěji dlouhodobě užívaných skupin léků, v roce 2011 bylo v České republice vydáno více než 350 milionů denních dávek statinů. Zohledníme-li neustále stoupající trend spotřeby, pak v současné době je
statiny léčen více než milion nemocných, tedy každý desátý, ve věku seniorů pak téměř každý druhý. Nejčastěji je užíván atorvastatin (59 %), simvastatin (21 %), rosuvastatin (16 %), fluvastatin (4 %) a necelé jedno procento spadá na lovastatin a pravastatin. Obdobná distribuce byla pozorována při analýze preskripce statinů v ordinacích ambulantních specialistů v České republice ve studii STEP [1]. Atorvastatin byl předepisován u 54 %, simvastatin u 21 %, rosuvastatin v 19 %, fluvastatin v 5 % a v necelém procentu statiny ostatní. Přitom se dá jednoznačně říci, že současný stav léčby dyslipidemie u nás není optimální, i v ordinacích ambulantních specialistů (kardiologů, internistů, lipidologů a diabetologů) je dosahováno cílových hodnot u méně než 50 % nemocných. Nejhorší situace je paradoxně u vysoce rizikových jedinců (po kardiovaskulární příhodě), kde je léčebným cílem pokles koncentrace LDL cholesterolu pod hranici 2,5 mmol/l; zde docilujeme cílové hodnoty dokonce jen ve 20 % [1]. Je možné výběrem statinu tuto situaci zlepšit? Porovnejme statiny z pohledu účinnosti a bezpečnosti.
Nejprve analyzujme statiny z pohledu farmakologického, tj. srovnejme farmakokinetické a farmakodynamické vlastnosti, posléze z pohledu klinického, tj. porovnejme účinnost a bezpečnost, a konečně z pohledu ekonomického, tj. porovnejme náklady a efektivitu.
Farmakologický pohled
Mechanismus účinku statinů je shodný – kompetitivní blokáda HMG-CoA (hydroxymethylglutaryl-koenzym A) reduktázy, „rate-limiting“ kroku syntézy sterolového jádra cholesterolu. Aktivní místo (farmakofor) všech statinů je v jádře stejné, má obdobnou strukturu jako HMG-CoA a s koenzymem soutěží o vazbu v místě katalytického místa reduktázy. Inhibice syntézy již na počátku řetězce je výhodná, nehromadí se toxický meziprodukt, nadbytečný acetyl CoA je zpracován v citrátovém cyklu. Snížení tvorby cholesterolu v hepatocytu (a mírně též v jiných tkáních, zejména ve svalu) vede ke snížení nabídky cholesterolu v tkáních. Intenzivní vychytávání esterů cholesterolu vázaného v nízkodenzitních lipoproteinech (LDL) a nižší koncentrace cholesterolu v aterogenních lipoproteinech produkovaných játry (VLDL transformovaných na IDL a LDL) snižuje hladinu cholesterolu v LDL.
Vzhledem k tomu, že inhibice je kompetitivní, je důležitá doba, po kterou je aktivita HMG-CoA reduktázy blokována. Po období, kdy již koncentrace statinu není dostatečná, dochází ke vzniku tzv. rebound-fenoménu. Tak je po odeznění inhibice HMG-CoA reduktáza zvýšeně aktivní a dojde k intenzivní syntéze chybějícího cholesterolu. Účinek statinu se tímto mechanismem snižuje. Paralelně s blokádou syntézy sterolového jádra je inhibována tvorba ubiquinonu (koenzymu Q10 potřebného k transportu přes biomembránu na mitochondriální úrovni) či dolicholu, geranylu a farnesylu (lipidických složek nutných k syntéze regulačních proteinů, resp. k jejich vazbě na buněčné membrány). Do jaké míry se podílí snížení nabídky těchto paralelních produktů na farmakologickém účinku a na nežádoucím působení, není zřejmé. Při porovnání je inhibice HMG-CoA reduktázy (při užití farmakoekvivalentních dávek statinů) srovnatelná, za rozdílnou účinností stojí zejména různá délka působení vedoucí ke zmíněnému rebound-fenoménu. Význam tzv. extralipidového účinku statinů není dořešen. Pro všechny molekuly máme laboratorní doklady o ovlivnění hemostázy, reparačních a proliferačních dějů, endoteliální dysfunkce a vazomotoriky či aterogeneze, nicméně naprostou většinu těchto pozorování můžeme vysvětlit též primárním působením na lipoproteinové spektrum. Jak lipoproteiny o nízké denzitě, tak vysokodenzitní lipoproteiny jsou významnými regulátory řady biologických pochodů. Doklad o tom, že by efekt některého statinu byl v tomto ohledu specifičtější, nemáme.
Význam rozdílů v absorpci a transformaci u jednotlivých statinů
Hla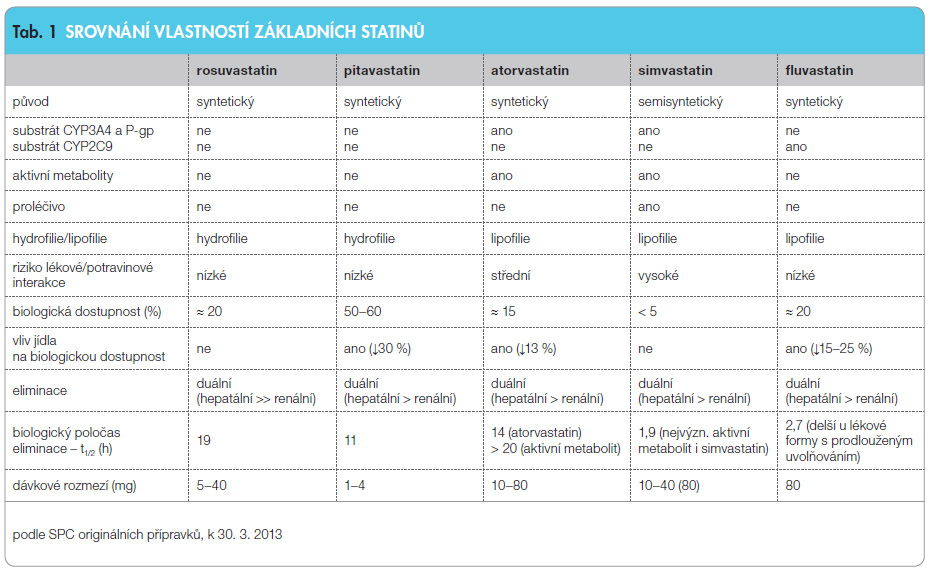 vní rozdělení na statiny lipofilní a hydrofilní má své opodstatnění. Lipofilní statiny podléhají během absorpce v enterocytu a při průchodu játry transformaci izoenzymy CYP, jejich dostupnost se tak snižuje a zvyšuje se riziko lékových interakcí. Stejně tak před eliminací musí být transformovány těmito oxidázami na hydrofilní metabolit a i zde dochází k interakci. Při seřazení podle lipofility se na vrcholu pomyslného žebříčku nachází cerivastatin a postupně klesá rozpustnost u lovastatinu, simvastatinu, fluvastatinu a atorvastatinu. Hydrofilní statiny rosuvastatin, pitavastatin a pravastatin nejsou významnými substráty metabolických systémů a více než 95 % je eliminováno jako mateřská látka. Srovnání vlastností základních statinů je uvedeno v tab. 1.
vní rozdělení na statiny lipofilní a hydrofilní má své opodstatnění. Lipofilní statiny podléhají během absorpce v enterocytu a při průchodu játry transformaci izoenzymy CYP, jejich dostupnost se tak snižuje a zvyšuje se riziko lékových interakcí. Stejně tak před eliminací musí být transformovány těmito oxidázami na hydrofilní metabolit a i zde dochází k interakci. Při seřazení podle lipofility se na vrcholu pomyslného žebříčku nachází cerivastatin a postupně klesá rozpustnost u lovastatinu, simvastatinu, fluvastatinu a atorvastatinu. Hydrofilní statiny rosuvastatin, pitavastatin a pravastatin nejsou významnými substráty metabolických systémů a více než 95 % je eliminováno jako mateřská látka. Srovnání vlastností základních statinů je uvedeno v tab. 1.
Způsob absorpce, distribuce, transformace (aktivace či inaktivace) a exkrece má zásadní význam pro výsledný interakční potenciál.
Simvastatin, lovastatin a atorvastatin jsou substráty eliminační pumpy glykoproteinu P (P-gp) a oxidázy CYP3A4/5. Tato dvojice tvoří funkční celek. U lipofilních molekul CYP3A4/5 katalyzuje oxidaci (vnesením hydrofilní hydroxylové skupiny). Tím je zvýšena afinita k P-gp a je umožněn eflux z buňky (z enterocytu do střeva, z hepatocytu do biliárního systému či z tubulární buňky nefronu do moče). Proto i exprese obou systémů je regulována stejným mechanismem – pregnanovým receptorem. Tak bývá aktivace i inhibice navozena stejnými molekulami – léky či složkami potravy. Tato vazba má svou logiku, tandem P-gp i CYP3A4/5 je hlavní bariérou zajišťující organismus před nadměrnou expozicí xenobiotikům, snižuje se absorpce, zvyšuje se eliminace. Vysoká afinita k transportnímu a metabolickému systému snižuje dostupnost dané molekuly a současně zvyšuje riziko interakce. Konkrétně u simvastatinu a lovastatinu je výsledná biologická dostupnost nízká, do systémového oběhu se dostává jen méně než 5 % léčiva, u atorvastatinu je biologická dostupnost vyšší, pohybuje se kolem 15 %.
![Graf 1 a–f Ovlivnění expozice statinům při jejich kombinaci s inhibitorem eliminačního a metabolického systému (P-gp a oxidázy CYP3A4/5); podle [3] – Jacobson, 2004. a, b, c) expoziční křivky pravastatinu, simvastatinu a atorvastatinu při podání samotného statinu a statinu v kombinaci se středně silným inhibitorem CYP 3A4 a P-gp – verapamilem; d, e, f) expoziční křivky pravastatinu, simvastatinu a atorvastatinu při podání samotného statinu a statinu v kombinaci se silným inhibitorem CYP 3A4 a P-gp – klarithromycinem; atorva – atorvastatin; klarit – klarithromycin; prava – pravastatin; simva – simvastatin; verap – verapamil](https://www.remedia.cz/photo-a-29293---.jpg) V kardiologické praxi se běžně setkáváme se současným podáváním statinů s verapamilem či s antiarytmiky (amiodaronem či propafenonem), tedy se středně silnými inhibitory obou systémů. Expozice (součin plazmatické koncentrace a délky působení) tak stoupá u simvastatinu při současném podání středně silného inhibitoru CYP3A4/5 a P-gp (v praxi nejčastěji verapamilu, propafenonu či amiodaronu) asi trojnásobně a při aplikaci silného inhibitoru (např. klarithromycinu či azolových antimykotik) více než desetinásobně. U atorvastatinu je vzestup expozice nižší, u středně silných inhibitorů jen dvojnásobný, u silných trojnásobný [2, 3]. Zvýšení expozice po podání středního a silného inhibitoru k léčbě lipofilními statiny (simvastatin a atorvastatin) či ke statinu hydrofilnímu (pravastatin) je ukázáno na průběhu expozičních křivek (graf 1 a–f). Podobně při současném podání induktorů obou systémů (řada antikonvulziv, dexamethason, hyperforin z extraktu třezalky či rifampin) snižuje expozici simvastatinu či lovastatinu na méně než 10 % [4]. Klinický dopad inhibice CYP3A4/5 u atorvastatinu je významně snížen skutečností, že tento enzym oxiduje molekulu atorvastatinu na ortho- a para-hydroxyderiváty, které jsou v inhibici HMG-CoA reduktázy stejně účinné jako mateřská molekula. Výpadek aktivity CYP3A4/5 má tak menší dopad na hypolipidemický efekt.
V kardiologické praxi se běžně setkáváme se současným podáváním statinů s verapamilem či s antiarytmiky (amiodaronem či propafenonem), tedy se středně silnými inhibitory obou systémů. Expozice (součin plazmatické koncentrace a délky působení) tak stoupá u simvastatinu při současném podání středně silného inhibitoru CYP3A4/5 a P-gp (v praxi nejčastěji verapamilu, propafenonu či amiodaronu) asi trojnásobně a při aplikaci silného inhibitoru (např. klarithromycinu či azolových antimykotik) více než desetinásobně. U atorvastatinu je vzestup expozice nižší, u středně silných inhibitorů jen dvojnásobný, u silných trojnásobný [2, 3]. Zvýšení expozice po podání středního a silného inhibitoru k léčbě lipofilními statiny (simvastatin a atorvastatin) či ke statinu hydrofilnímu (pravastatin) je ukázáno na průběhu expozičních křivek (graf 1 a–f). Podobně při současném podání induktorů obou systémů (řada antikonvulziv, dexamethason, hyperforin z extraktu třezalky či rifampin) snižuje expozici simvastatinu či lovastatinu na méně než 10 % [4]. Klinický dopad inhibice CYP3A4/5 u atorvastatinu je významně snížen skutečností, že tento enzym oxiduje molekulu atorvastatinu na ortho- a para-hydroxyderiváty, které jsou v inhibici HMG-CoA reduktázy stejně účinné jako mateřská molekula. Výpadek aktivity CYP3A4/5 má tak menší dopad na hypolipidemický efekt.
Význam změny aktivity P-gp na základě polymorfismu ABCB1 (řídícího genu této pumpy) se projeví jen s hraniční významností, expozice atorvastatinu či simvastatinu se při nízké aktivitě pumpy zvýšila jen o 50 % [5].
Fluvastatin je velmi slabým substrátem P-gp a středně silným substrátem a současně inhibitorem izoenzymu 2C9. Tato oxidáza je polymorfní, rozlišujeme metabolizátory rychlé (se sníženou expozicí fluvastatinu), střední (se standardní expozicí) a pomalé (s expozicí vyšší). Tyto rozdíly mohou být klinicky významné – rozdíly v účinku fluvastatinu byly sledovány v české populaci – u rychlých metabolizátorů byla hladina LDL cholesterolu snížena o 22,4 %, u středních o 25 % a u pomalých o 40 % [6]. Doklady o tom, že by farmakogenetické vlivy či lékové interakce na této úrovni významně ovlivnily hypolipidemický účinek či výskyt myalgií/myopatií při léčbě fluvastatinem,
nemáme.
Podívejme se i na druhou stranu, tedy na inhibici CYP2C9 fluvastatinem. Jak bylo řečeno, fluvastatin tuto oxidázu středně silně inhibuje a tak může ovlivnit metabolismus jiných léků. Nejvýznamnějším substrátem izoenzymu CYP2C9 je warfarin, lék s velmi úzkým terapeutickým oknem. Zvýšení expozice warfarinu je středně významné, vzestup hladiny warfarinu činil 30–40 %, resp. hodnota INR (international normalized ratio) se zvýšila o 0,5 až 1 a dávku warfarinu je často nutno snižovat [7]. Rosuvastatin, pravastatin a pitavastatin nejsou významnými substráty oxidáz CYP, jejich aktivitu ani neovlivňují.
Význam rozdílů v distribuci a eliminaci
Druhým klíčovým momentem je transport statinů (lipofilních i hydrofilních) do hepatocytu, tedy do místa vlastní inhibice HMG-CoA reduktázy. Transmembranózní přenos je výrazně facilitován transportní pumpou OATP1B1 (organic aniont transporting polypeptid 1B1). Na krevním pólu hepatocytu tento protein umožní vstup statinu do hepatocytu, na biliárním pólu pak další pumpy (zejména P-gp) umožní eflux do žluče (obr. 1). Nedostatečný transport do jaterní buňky vede k nižší koncentraci statinu na jaterních ribozomech a ke snížené inhibici HMG-CoA reduktázy. Plazmatická koncentrace statinu je při zpomalené exkreci biliárním systémem zvýšena, narůstá i frekvence nežádoucích účinků. Závislost vstupu jednotlivých statinů do hepatocytu na aktivitě OATP1B1 je různá, nejvíce j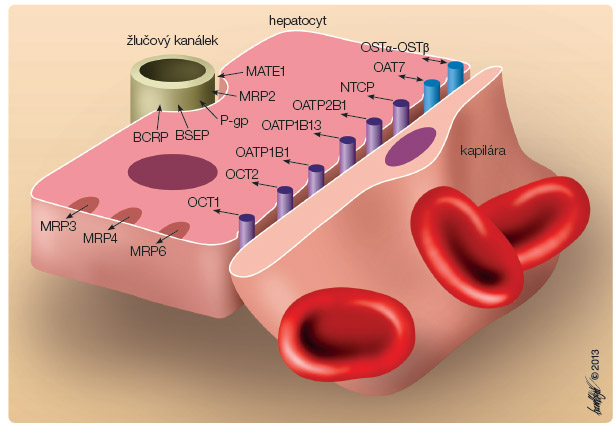 e stimulován vstup simvastatinu, nejméně fluvastatinu a rosuvastatinu (simva- > pitava- > atorva- > prava- > rosuva- > fluvastatin) [8, 9].
e stimulován vstup simvastatinu, nejméně fluvastatinu a rosuvastatinu (simva- > pitava- > atorva- > prava- > rosuva- > fluvastatin) [8, 9].
Mnohé z léků často podávaných současně se statiny (fibráty, dipyridamol, sartany, zejména telmisartan a kandesartan, či nesteroidní antirevmatika) významně inhibují aktivitu této pumpy a zvyšují plazmatickou koncentraci statinů – nejvíce v případě simvastatinu (o 200–300 %), nejméně u fluvastatinu a rosuvastatinu (o 20–70 %) [8, 9]. Zvýšení rizika výskytu myopatií při současné léčbě statiny (zejména cerivastatinem a simvastatinem) s fibráty, zejména s gemfibrozilem, je dobře známo, menší povědomí je o tomto typu interakce v souvislosti s léčbou sartany či nesteroidními antiflogistiky [10].
Nabí![Graf 2 Změna expozice statinům u varianty genotypu se sníženou nebo nulovou aktivitou transportéru OATP1B1; podle [11] – Voora, et al., 2009. atorva – atorvastatin; fluva – fluvastatin; prava – pravastatin; rosuva – rosuvastatin; simva – simvastatin](https://www.remedia.cz/photo-a-29295---.jpg) dka pumpy OATP1B1 je řízena genem SLCO1B1. V populaci se vyskytuje variace genu SLCO1B1 (varianta rs4149056) se sníženou aktivitou pumpy typu „loss-of-function“. V heterozygotní formě (alely TC) tento polymorfismus, vyskytující asi u 15 % indoevropské populace, významně snižuje aktivitu transportéru a v homozygotní formě (alely CC), nacházené asi u 2 % populace, je aktivita nulová. Nízká aktivita influxní/efluxní pumpy OATP1B1 je klinicky významná, díky snížené eliminaci do žluče se zvyšuje plazmatická koncentrace statinů. Zvýšení expozice je nejvíce vyjádřeno u simvastatinu (graf 2) [8].
dka pumpy OATP1B1 je řízena genem SLCO1B1. V populaci se vyskytuje variace genu SLCO1B1 (varianta rs4149056) se sníženou aktivitou pumpy typu „loss-of-function“. V heterozygotní formě (alely TC) tento polymorfismus, vyskytující asi u 15 % indoevropské populace, významně snižuje aktivitu transportéru a v homozygotní formě (alely CC), nacházené asi u 2 % populace, je aktivita nulová. Nízká aktivita influxní/efluxní pumpy OATP1B1 je klinicky významná, díky snížené eliminaci do žluče se zvyšuje plazmatická koncentrace statinů. Zvýšení expozice je nejvíce vyjádřeno u simvastatinu (graf 2) [8].
Paralelně se vzestupem hladiny statinů v plazmě byl doložen výrazně vyšší výskyt myalgií (bolestivost svalstva bez v![Graf 3 Zvýšení výskytu myopatií u genotypu s nulovou aktivitou transportéru OATP1B1 (homozygot s inaktivními alelami – varianta CC), s aktivitou sníženou (heterozygot s jednou inaktivní alelou – varianta TC) ve srovnání s běžným genotypem (obě alely TT); podle [11] – Voora, et al., 2009.](https://www.remedia.cz/photo-a-29296---.jpg) zestupu hladin kreatinkinázy, CK) a myopatií (již se vzestupem CK). Ve farmakogenetické studii SEARCH bylo sledováno po dobu 6 let riziko výskytu nežádoucích účinků u 32 tisíc nemocných léčených statiny, z toho 20 tisíc bylo léčeno simvastatinem [11, 12]. U heterozygotů s polymorfismem typu loss-of-function (genotyp TC) léčených simvastatinem ve vyšší dávce došlo ke zvýšení výskytu myopatií pětinásobně (HR 4,5; 95% CI: 2,6–7,7), u homozygotů (alely CC) bylo zvýšení 17násobné (HR 16,9; 95% CI: 4,7– 61,1). Při srovnání frekvence významných a ověřených myopatií stoupla frekvence u homozygotů téměř třicetinásobně (HR 27,2; 95% CI: 6,8–109,2), viz graf 3. Přítomnost varianty s nízkou či nulovou aktivitou pumpy OATP1B1 vysvětluje asi 60 % pozorovaných myopatií.
zestupu hladin kreatinkinázy, CK) a myopatií (již se vzestupem CK). Ve farmakogenetické studii SEARCH bylo sledováno po dobu 6 let riziko výskytu nežádoucích účinků u 32 tisíc nemocných léčených statiny, z toho 20 tisíc bylo léčeno simvastatinem [11, 12]. U heterozygotů s polymorfismem typu loss-of-function (genotyp TC) léčených simvastatinem ve vyšší dávce došlo ke zvýšení výskytu myopatií pětinásobně (HR 4,5; 95% CI: 2,6–7,7), u homozygotů (alely CC) bylo zvýšení 17násobné (HR 16,9; 95% CI: 4,7– 61,1). Při srovnání frekvence významných a ověřených myopatií stoupla frekvence u homozygotů téměř třicetinásobně (HR 27,2; 95% CI: 6,8–109,2), viz graf 3. Přítomnost varianty s nízkou či nulovou aktivitou pumpy OATP1B1 vysvětluje asi 60 % pozorovaných myopatií.
Tato studie není ojedinělá, ke stejnému závěru dospěla retrospektivní farmakogenetická analýza probandů léčených simvastatinem v Heart Protection Study. Výskyt myalgie/myopatie byl rovněž několikanásobně zvýšen u nositelů alely TC či CC [13]. Podobně metaanalýza farmakogenetických studií sledujících rizikové faktory rhabdomyolýzy ukazuje, že popsaný polymorfismus OATP1B1 typu loss-of-function je pro vznik závažných myopatií faktorem rozhodujícím [14]. Varianty měnící aktivitu ostatních transportních a metabolických systémů měly význam podstatně menší.
Dopad snížené aktivity OATP1B1 byl u ostatních statinů významně nižší. Transport atorvastatinu a pravastatinu do hepatocytu je rovněž stimulován tímto transportérem, penetrace do hepatocytu je umožněna i jinými mechanismy. Ve studii STRENGHT byl porovnáván efekt přítomnosti alel TC a CC na intoleranci a výskyt nežádoucích účinků též u ostatních statinů, u simvastatinu, atorvastatinu a pravastatinu [15]. Pro simvastatin byla asociace největší, riziko se zvyšovalo trojnásobně pro každou kopii alely C, u atorvastatinu byl dopad menší, riziko nežádoucích účinků vzrostlo jen o 60 %, pro pravastatin vazba pozorována nebyla. Podobné, násobné zvýšení rizika u nemocných s výše uvedeným polymorfismem při léčbě simvastatinem, nikoli však atorvastatinem, nacházejí i ostatní autoři [9].
Na základě významné závislosti účinnosti a bezpečnosti simvastatinu na farmakogenetické výbavě a s ohledem na zvýšený výskyt myopatií při léčbě vyššími dávkami simvastatinu bylo vydáno varování Food and Drug Administration (FDA) před užíváním vyšších dávek (80 mg) simvastatinu v léčbě všech nemocných s výjimkou těch, kteří již dlouhodobě tuto dávku dobře tolerovali před zveřejněním „black boxu“. Dávku 40 mg FDA nedoporučuje při současném podávání středně silných či slabých inhibitorů P-gp/CYP3A4/5 či OATP1B1 (jmenovitě amiodaronu či amlodipinu) a ani dávku 20 mg nepokládá za vhodnou při léčbě silnými a středně silnými inhibitory (opět jmenovitě např. při podávání verapamilu či diltiazemu). Obecně doporučuje preferovat volbu alternativního statinu.
Konsorcium zástupců Společnosti klinické farmakogenomiky vydává pro léčbu simvastatinem doporučený postup [9]. Zde konstatuje obecně dobrou toleranci statinů, v metaanalýze studií nachází výskyt myalgií v 1–5 %, na každých 17 léčených se objevuje v průměru jeden případ. Myopatie je podstatně méně častá, analýza uvádí výskyt ~ 1 : 1000
a pro rhabdomyolýzu incidenci ~ 1 : 1000 000.
U simvastatinu tato metaanalýza prokazuje zvýšení incidence nad tento průměr. Přítomnost polymorfismu SLCO1B1 typu loss-of-function v heterozygotní formě (alely TC) zvyšuje riziko myopatie 4–5násobně a v homozygotní formě asi dvacetinásobně. Shrneme-li, pak po stránce farmakokinetiky existují významné rozdíly mezi jednotlivými molekulami statinů. Klinický význam má jistě zvýšení hladiny simvastatinu (a obdobně lovastatinu) při inhibici P-gp a CYP3A4/5 a při variantě genomu typu loss-of-function. Zvýšení rizika vzniku myalgií, myopatií až rhabdomyolýzy dokumentuje řada studií a tato skutečnost je zohledněna i ve varování regulačních orgánů a v doporučených postupech. Klinický význam lékových interakcí a farmakogenetických variant u ostatních statinů (včetně atorvastatinu) je podstatně menší.
Klinický pohled
Pro klinika je důležitá zejména účinnost v užívaných indikacích, snášenlivost a bezpečnost. Indikace jsou u všech statinů stejné: snížení hladiny LDL cholesterolu k cílovým hodnotám v rámci primární a sekundární prevence z důvodu snížení kardiovaskulární morbidity a mortality. Některé statiny mají navíc schváleno podávání ve specifických situacích, např. pravastatin u nemocných léčených imunosupresivy po transplantaci orgánů. Podávání jiných (např. fluvastatinu) je schváleno u dětí a adolescentů s familiární hypercholesterolemií, nicméně i zde je praxe v jednotlivých zemích různá. Zásadní rozdíly ve schválených indikacích však nejsou, v tomto bodě se statiny významně neliší.
Rozdíly v účinnosti
Srovnáme-li jednotlivé statiny z pohledu účinnosti, tedy základního účinku – redukce hladiny LDL cholesterolu, v![Graf 4 Srovnání poklesu plazmatické koncentrace LDL cholesterolu při léčbě třemi nejčastěji užívanými statiny; podle [16] – Jones, et al., 2003.](https://www.remedia.cz/photo-a-29297---.jpg) zestupu hodnot HDL cholesterolu a eventuálního vlivu na triacylglyceroly (triglyceridy) – jsou mezi statiny významné rozdíly. Efekt na pokles hladin LDL cholesterolu a jejich vzestup u HDL cholesterolu není překvapivě paralelní, není ani obdobný u všech statinů, a dokonce není ani stejný u různých dávek téhož statinu. Při analýze vlivu na pokles hladin LDL cholesterolu jsou nejpotentnější rosuvastatin, atorvastatin a pitavastatin [16–18]. Rosuvastatin ve středních dávkách, atorvastatin a pitavastatin v nejvyšší dávce jsou schopny snížit hladinu LDL cholesterolu o více než
zestupu hodnot HDL cholesterolu a eventuálního vlivu na triacylglyceroly (triglyceridy) – jsou mezi statiny významné rozdíly. Efekt na pokles hladin LDL cholesterolu a jejich vzestup u HDL cholesterolu není překvapivě paralelní, není ani obdobný u všech statinů, a dokonce není ani stejný u různých dávek téhož statinu. Při analýze vlivu na pokles hladin LDL cholesterolu jsou nejpotentnější rosuvastatin, atorvastatin a pitavastatin [16–18]. Rosuvastatin ve středních dávkách, atorvastatin a pitavastatin v nejvyšší dávce jsou schopny snížit hladinu LDL cholesterolu o více než 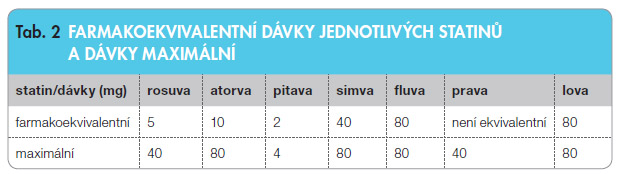 polovinu. Simvastatin, fluvastatin, pravastatin a lovastatin jsou i při podání nejvyšších dávek méně účinné (graf 4). Porovnání farmakoekvivalentních a maximálních dávek je uvedeno v tab. 2.
polovinu. Simvastatin, fluvastatin, pravastatin a lovastatin jsou i při podání nejvyšších dávek méně účinné (graf 4). Porovnání farmakoekvivalentních a maximálních dávek je uvedeno v tab. 2.
Při analýze dopadu jednotlivých statinů na koncentraci HDL cholesterolu je doložen mírně vyšší efekt vyšších dávek rosuvastatinu na vzestup hladiny (asi 5–10 %), u ostatních statinů se pohybuje vzestup kolem 2–5 % a pouze vyšší dávky atorvastatinu hladinu HDL cholesterolu nezvyšují (graf 5). Zda js![Graf 5 Srovnání vzestupu plazmatické koncentrace HDL cholesterolu při léčbě třemi nejčastěji užívanými statiny; podle [16] – Jones, et al., 2003.](https://www.remedia.cz/photo-a-29299---.jpg) ou tyto rozdíly v ovlivnění ateroprotektivního lipoproteinu klinicky významné, nevíme. Pokles triglyceridemie je při podávání všech statinů (při srovnání farmakoekvivalentních dávek) srovnatelný [18, 6]. Dávka 10 mg rosuvastatinu, 4 mg pitavastatinu, 20 mg atorvastatinu a 80 mg simvastatinu či fluvastatinu snižuje hladinu triacylglycerolu asi o 20 %.
ou tyto rozdíly v ovlivnění ateroprotektivního lipoproteinu klinicky významné, nevíme. Pokles triglyceridemie je při podávání všech statinů (při srovnání farmakoekvivalentních dávek) srovnatelný [18, 6]. Dávka 10 mg rosuvastatinu, 4 mg pitavastatinu, 20 mg atorvastatinu a 80 mg simvastatinu či fluvastatinu snižuje hladinu triacylglycerolu asi o 20 %.
Rozdíly v klinickém účinku, tj. v poklesu mortality/morbidity, nelze porovnávat. U jednotlivých statinů nejsou k dispozici obdobně koncipované studie – nejen pro analýzu „head to head“, ale ani pro síťovou analýzu s nepřímým porovnáním vůči placebu. Při porovnávání efektu dvou statinů byly studie vždy koncipovány k doložení většího účinku intenzivnější léčby vysokou dávkou srovnávaného statinu se základní či běžnou dávkou komparátoru. Porovnání prognostického efektu jednotlivých statinů s placebem v základních indikacích máme jen pro prvou generaci statinů, pro další generace by obdobné studie byly již neetické. Shrneme-li, pak efekt jednotlivých statinů na lipidové spektrum je odlišný, při použití farmakoekvivalentních základních dávek však klinický význam není veliký. Zásadní rozdíl můžeme pozorovat pouze při užití vyšších dávek potentnějších statinů (rosuvastatinu, atorvastatinu či pitavastatinu), zde docílíme poklesu hladiny LDL cholesterolu řádově o 50 %. Při užití maximálních dávek méně účinných statinů (simvastatinu, fluvastatinu, pravastatinu či lovastatinu) nedocílíme poklesu většího než o 30–40 %. Tomu odpovídá i nižší procento nemocných, kteří dosáhnou cílové hladiny LDL cholesterolu při užití této kategorie statinů.
Rozdíly v bezpečnosti a toleranci
Statiny jsou jednou z nejlépe tolerovaných a nejbezpečnějších lékových skupin. Nicméně, vzhledem k tomu, že je jimi léčena asi desetina populace, má výskyt i nežádoucích účinků snižujících pouze kvalitu života velký význam. Vedou totiž ke snížení spolupráce pacienta, k redukci dávky či k ukončení léčby statinem. Nejčastěji se vyskytující nežádoucí účinek (myalgie) není sám o sobě nebezpečný, nicméně přerušení či ukončení léčby může nemocného připravit o přínos spojený s léčbou dyslipidemie a ohrozit jej zvýšením kardiovaskulárního rizika.
Jak bylo zdůrazněno, závažné nežádoucí účinky statinů jsou vzácné (tab. 3). Riziko výskytu však je vyšší u seniorů, žen, o sob s nižší hmotností, u osob s vyšší fyzickou aktivitou, s metabolickými poruchami (např. při hypotyreóze) či při renální či hepatální nedostatečnosti. Podle statistik regulačních orgánů (zejména FDA) frekvence výskytu myalgií, myopatií a myolýzy několikanásobně stoupá při současném podávání jiných léků, jako příčina se uvádí zvýšení expozice statinu na podkladě lékových interakcí s inhibitory metabolických a transportních systémů.Bylo doloženo, že riziko lékových interakcí a genetických polymorfismů ovlivňujících expozici i výskyt nežádoucích účinků je u jednotlivých statinů odlišné. Zásadní otázka tedy zní: Jsou tyto rozdíly klinicky významné a odrazily se poznatky ze sledování farmakokinetiky a farmakogenetických studií v toleranci léčby? Překvapivě málo studií je zaměřeno na rozdíly ve výskytu nežádoucích účinků, konkrétně myalgií a myopatií při léčbě statiny.
sob s nižší hmotností, u osob s vyšší fyzickou aktivitou, s metabolickými poruchami (např. při hypotyreóze) či při renální či hepatální nedostatečnosti. Podle statistik regulačních orgánů (zejména FDA) frekvence výskytu myalgií, myopatií a myolýzy několikanásobně stoupá při současném podávání jiných léků, jako příčina se uvádí zvýšení expozice statinu na podkladě lékových interakcí s inhibitory metabolických a transportních systémů.Bylo doloženo, že riziko lékových interakcí a genetických polymorfismů ovlivňujících expozici i výskyt nežádoucích účinků je u jednotlivých statinů odlišné. Zásadní otázka tedy zní: Jsou tyto rozdíly klinicky významné a odrazily se poznatky ze sledování farmakokinetiky a farmakogenetických studií v toleranci léčby? Překvapivě málo studií je zaměřeno na rozdíly ve výskytu nežádoucích účinků, konkrétně myalgií a myopatií při léčbě statiny.
Studie PRIMO je z metodického hlediska k porovnání nejvhodnější, neboť monitorovala výskyt myalgií/myopatií v reálné praxi [19]. Z osmi tisíc probandů léčených statiny (simva-, atorva-, prava- a fluvastatinem) se myalgie/![Graf 6 Výskyt myalgií/myopatií při léčbě vyššími dávkami statinů ve studii PRIMO; podle [19] – Bruckert, 2005. atorva – atorvastatin; fluva – fluvastatin; prava – pravastatin; simva – simvastatin](https://www.remedia.cz/photo-a-29301---.jpg) myopatie objevily u 10 % z nich. Při léčbě simvastatinem (v dávce 40–80 mg denně) se myalgie/myopatie objevují v 18,2 %, v porovnání s fluvastatinem (v dávce 80 mg denně) byly téměř čtyřikrát častější (graf 6). Rozdíly ve výskytu svalových potíží byly při léčbě simvastatinem i atorvastatinem (40 až 80 mg denně) významné také v porovnání s pravastatinem (40 mg denně) či s fluvastatinem. Dávky užité při podávání simvastatinu a fluvastatinu přitom byly z hlediska účinnosti na pokles hladiny LDL cholesterolu přibližně ekvivalentní, naopak obě dávky atorvastatinu byly významně účinnější.
myopatie objevily u 10 % z nich. Při léčbě simvastatinem (v dávce 40–80 mg denně) se myalgie/myopatie objevují v 18,2 %, v porovnání s fluvastatinem (v dávce 80 mg denně) byly téměř čtyřikrát častější (graf 6). Rozdíly ve výskytu svalových potíží byly při léčbě simvastatinem i atorvastatinem (40 až 80 mg denně) významné také v porovnání s pravastatinem (40 mg denně) či s fluvastatinem. Dávky užité při podávání simvastatinu a fluvastatinu přitom byly z hlediska účinnosti na pokles hladiny LDL cholesterolu přibližně ekvivalentní, naopak obě dávky atorvastatinu byly významně účinnější.
Jiná analýza se zabývala nežádoucími účinky při léčbě různými statiny (atorva-, rosuva-, prava- a simvastatinem) na základě dat klinických studií [20]. Při podávání výrazně nižších dávek statinů nebyly zjištěny rozdíly ve výskytu různých nežádoucích účinků. Důvodem je zřejmě jejich nízký výskyt v této analýze, myalgie se např. objevovaly jen ve 2–3 %.
Při sledování ostatních nežádoucích účinků, např. vzestupu hodnot jaterních testů či mírného zvýšení rizika manifestace diabetu, se statiny mezi sebou neliší [21, 22]. Například riziko vzniku diabetu se pohybovalo při léčbě všemi statiny kolem zvýšení incidence o jeden případ na tisíc léčených po dobu jednoho roku. Tedy riziko bylo významně nižší, než s jakým se setkáváme např. při léčbě diuretiky. Rovněž riziko zvýšení hodnot jaterních testů nemá velký význam, např. FDA inovovala svůj názor a upustila od doporučení pravidelných laboratorních kontrol v průběhu léčby statiny.
Shrneme-li, pak rozdíly v toleranci statinů (zejména pro výskyt myalgií/myopatií) při užití nižších dávek nemají zásadní klinický význam. Je však doloženo, že při podávání vyšších dávek statinů je fluvastatin nejbezpečnější (za cenu nižší účinnosti na pokles hladin LDL cholesterolu) a simvastatin je zatížen naopak nejvýraznějším výskytem myalgií a myopatií. U atorvastatinu a rosuvastatinu, ač patří k nejúčinnějším statinům, není léčba spojena s vyšším výskytem myalgií/myopatií či jiných nežádoucích účinků ve srovnání se statiny méně účinnými.
Farmakoekonomický pohled
Po stránce farmakoekonomické splňují statiny i ta nejpřísnější kritéria efektivity. Platí to pro všechny zavedené indikace, tj. profylaxi kardiovaskulárních příhod a úmrtí v rámci primární prevence u rizikových osob či jejich užití v rámci sekundární prevence. Statiny jsou vysoce účinné (v metaanalýzách je doložen pokles mortality/morbidity v řádu 20–30 %), bezpečné (život ohrožující komplikace se objevují řádově s frekvencí jedno úmrtí na jeden milion léčených ročně) a nízké jsou i finanční náklady: cena denní dávky se pohybuje kolem 3–6 Kč. Při porovnání statinů je cena i úhrada farmakoekvivalentních dávek statinů srovnatelná, významné rozdíly nenalezneme.
Při výběru nejvhodnějšího statinu pro daného nemocného bychom měli přihlížet k účinnosti, bezpečnosti a snášenlivosti. Náklady na léčbu, tedy na obdobný pokles LDL cholesterolu, jsou u statinů dostupných v ČR srovnatelné. Porovnat vzájemně efekt na pokles mortality a morbidity neumíme, dosavadní studie srovnávaly statiny vždy jen z pohledu intenzivnější léčby s léčbou standardní, nikoli při aplikaci dávek ekvivalentních. Rosuvastatin a atorvastatin ve vyšších dávkách mají doložen největší pokles hladiny LDL cholesterolu, tyto preferujeme všude tam, kde chceme dosáhnout poklesu LDL cholesterolu řádově o polovinu. U nemocných se současně nízkou hladinou HDL cholesterolu je nejvýhodnější rosuvastatin, vzestup hladiny v průměru o 5–10 % je srovnatelný se vzestupem po podávání fibrátů.
Druhé hledisko, tedy bezpečnost, je do velké míry závislé na výskytu variabilní expozice některým lipofilním statinům, zejména simvastatinu. Při užití vyšších dávek (u simvastatinu 40–80 mg/den) je bezpečnost léčby simvastatinem nižší. Stejně tak tolerance, zejména výskyt myalgií, je při užití vyšších dávek simvastatinu vyšší. U ostatních statinů je výskyt nežádoucích účinků srovnatelný, pouze podávání fluvastatinu je provázeno nejnižším výskytem myalgií a myopatií.
S bezpečností a snášenlivostí souvisí i hledisko lékových interakcí. Všude tam, kde nemocného současně léčíme inhibitory oxidázy CYP3A4/5 či inhibitory transportéru P-gp, tj. v kardiologii nejčastěji verapamilem, diltiazemem či amiodaronem, bychom neměli podávat statiny, které jsou substráty těchto systémů. Konkrétně bychom neměli volit simvastatin, lovastatin a vyšší dávky atorvastatinu. U nemocných léčených warfarinem naopak není výhodné volit primárně fluvastatin, který je středně silným inhibitorem biodegradace warfarinu. Pokud jej přece jen zvolíme, musíme po dobu zavádění léčby zintenzivnit kontroly INR.
Z praktického hlediska je vhodné při výběru vzít v úvahu též spolupráci nemocného. Pokud pacient vynechává léčebné dávky častěji, je výhodný rosuvastatin, u něhož jedno- či dvoudenní vynechání nevede k úplnému vymizení efektu. Po vynechání dávky na 48 hodin přetrvává ještě nejméně polovina hypolipidemického účinku.
Závěrem je nutno zdůraznit, že rozdíly mezi statiny nejsou zásadní. Daleko významnější je výběr vhodného nemocného a vhodné dávky. Stále ještě velká část indikovaných nemocných léčena není, a pokud léčena je, velmi často nedosahujeme optimálních cílových hodnot.
Seznam použité literatury
- [1] Hradec J, Bultas J, Kmínek A, et al. Jak se léčí statiny v České republice? Výsledky průzkumu STEP. Cor Vasa 2011; 53: 527–534.
- [2] Vaquero MP, Sánchez Muniz FJ, et al. Major diet-drug interactions affecting the kinetic characteristics and hypolipidaemic properties of statins. Nutr Hosp 2010; 25: 193–206.
- [3] Jacobson TA. Comparative pharmacokinetic interaction profiles of pravastatin, simvastatin, and atorvastatin when coadministered with cytochrome P450 inhibitors. Amer J Cardiol 2004; 94: 1140–1146.
- [4] Kyrklund C, Backman JT, Kivistö KT, et al. Rifampin greatly reduces plasma simvastatin and simvastatin acid concentrations. Clin Pharmacol Ther 2000; 68: 592–597.
- [5] Keskitalo JE, Kurkinen KJ, Neuvonen PJ, Niemi M. ABCB1 haplotypes differentially affect the pharmacokinetics of the acid and lactone forms of simvastatin and atorvastatin. Clin Pharmacol Ther 2008; 84: 457–461.
- [6] Buzková H, Pechandová K, Danzig V, et al. Lipid-lowering effect of fluvastatin in relation to cytochrome P450 2C9 variant alleles frequently distributed in the Czech population. Med Sci Monit 2012; 18: CR512–517.
- [7] Kim MJ, Nafziger AN, Kashuba AD, et al. Effects of fluvastatin and cigarette smoking on CYP2C9 activity measured using the probe S-warfarin. Eur J Clin Pharmacol 2006; 62: 431–436.
- [8] Niemi M, Pasanen MK, Neuvonen PJ. Organic anion transporting polypeptide 1B1: a genetically polymorphic transporter of major importance for hepatic drug uptake. Pharmacol Rev 2011; 63: 157–181.
- [9] Wilke RA, Ramsey LB, Johnson SG, et al., and Clinical Pharmacogenomics Implementation Consortium (CPIC). The clinical pharmacogenomics implementation consortium: CPIC guideline for SLCO1B1 and simvastatin-induced myopathy. Clin Pharmacol Ther 2012; 92: 112–117.
- [10] Kalliokoski A, Niemi M. Impact of OATP transporters on pharmacokinetics. Br J Pharmacol 2009; 158: 693–705.
- [11] Voora D, Shah SH, Spasojevic I, et al. The SLCO1B1*5 genetic variant is associated with statin-induced side effects. J Am Coll Cardiol 2009; 54: 1609–1616.
- [12] SEARCH Collaborative Group, Link E, Parish S, Armitage J, et al. SLCO1B1 variants and statin-induced myopathy – a genomewide study. N Engl J Med 2008; 359: 789–799.
- [13] Heart Protection Study Collaborative Group, Bulbulia R, Bowman L, Wallendszus K, et al. Effects on 11-year mortality and morbidity of lowering LDL cholesterol with simvastatin for about 5 years in 20,536 high-risk individuals: a randomised controlled trial. Lancet 2011; 378: 2013–2020.
- [14] Chiba K, Morimoto K. Genetic marker of statin-induced rhabdomyolysis. Yakugaku Zasshi 2011; 131: 247–253.
- [15] Puccetti L, Ciani F, Auteri A. Genetic involvement in statins induced myopathy. Preliminary data from an observational case-control study. Atherosclerosis 2010; 211: 28–29.
- [16] Jones PH, Davidson MH, Stein EA et STELLAR Study Group. Comparison of the efficacy and safety of rosuvastatin versus atorvastatin, simvastatin, and pravastatin across doses (STELLAR Trial). Am J Cardiol 2003; 92: 152–160.
- [17] Deedwania PC, Hunninghake DB, Bays HE, et al. Effects of rosuvastatin, atorvastatin, simvastatin, and pravastatin on atherogenic dyslipidemia in patients with characteristics of the metabolic syndrome. Am J Cardiol 2005; 95: 360–366.
- [18] Ose L. Pitavastatin: finding its place in therapy. Ther Adv Chronic Dis 2011; 2: 101–117.
- [19] Bruckert E, Hayem G, Dejager S, et al. Mild to moderate muscular symptoms with high-dosage statin therapy in hyperlipidemic patients-the PRIMO study. Cardiovasc Drugs Ther 2005; 19: 403–414.
- [20] Shepherd J, Vidt DG, Miller E, et al. Safety of rosuvastatin: update on 16,876 rosuvastatin-treated patients in a multinational clinical trial program. Cardiology 2007; 107: 433–443.
- [21] Koh KK, Quon MJ, Han SH, et al. Atorvastatin causes insulin resistance and increases ambient glycemia in hypercholesterolemic patients. Am Coll Cardiol 2010; 55: 1209–2016.
- [22] Sattar N, Preiss D, Murray HM, et al. Statins and risk of incident diabetes: a collaborative meta-analysis of randomised statin trials. Lancet 2010; 375: 735–742.