Získaná hemofilie A – diagnóza a současné možnosti farmakoterapie
Souhrn:
Získaná hemofilie A je vzácná forma získané koagulopatie způsobená autoprotilátkami proti koagulačnímu faktoru VIII. Je charakterizována závažnými, často život ohrožujícími krvácivými komplikacemi zejména u starších pacientů a u žen po porodu. Diagnóza je založena na klinickém obrazu a laboratorním vyšetření (prodloužený aktivovaný parciální tromboplastinový test, snížená aktivita koagulačního faktoru VIII a průkaz jeho specifického inhibitoru). Článek je zaměřen na klinické projevy, diagnostické a léčebné postupy s cílem zvýšit povědomí o tomto onemocnění mezi klinickými lékaři širokého spektra specializací.
Key words: acquired haemophilia – bleeding – bypassing agents – inhibitor eradication.
Summary:
Acquired haemophilia A is a rare form of the acquired coagulopathy caused by autoantibodies against coagulation factor VIII. It is characterized by major bleeding complications, often life-threatening, especially in elderly patients and postpartum women. Diagnosis is based on the clinical features and presence of a prolonged activated partial thromboplastin test, reduced activity of the coagulation factor VIII, and positive specific inhibitor assay. This article focuses on the clinical aspects, diagnostic and therapeutic procedures with the aim to increase awareness of this disorder among clinicians in a wide range of specialties.
Úvod
Získaná hemofilie A (acquired haemophilia A, AHA) je autoimunitní onemocnění projevující se často nečekaně závažným krvácením u mužů i žen s dosud negativní anamnézou krvácivého onemocnění v osobní či rodinné anamnéze a neužívajících antitrombotické či antikoagulační léky. S klasickou vrozenou hemofilií A s gonosomálně recesivní dědičností má společnou pouze nedostatečnou aktivitu koagulačního faktoru FVIII (FVIII:C). Etiologie, krvácivé projevy i léčba AHA jsou odlišné.
Epidemiologie a patogeneze získané hemofilie A
Odhadovaná roční incidence AHA je přibližně 1‒2 případy na milion obyvatel, obvykle postihuje starší pacienty bez rozdílu pohlaví ve věkovém mediánu 75‒80 let, v době diagnózy je pouze 15 % pacientů mladších 65 let [1,2]. U mladých nemocných ve věku 20‒40 let dominuje výskyt u žen v souvislosti s graviditou. U pacientek s postpartální AHA sledovaných v dosud nejrozsáhlejším prospektivním registru EACH2 (European Acquired Haemophilia Registry) se první krvácivá epizoda objevila v mediánu 2,5 měsíce po porodu, nicméně u 19 % z nich se příznaky korelující s diagnózou AHA objevily již před porodem [3]. U gravidních žen s diagnózou AHA hrozí riziko transplacentárního přenosu protilátek inhibujících aktivitu FVIII také na plod, krvácivé komplikace se mohou vyskytnout i u novorozence. Kromě gravidity jsou dalšími rizikovými faktory vzniku AHA autoimunitní choroby (zejména lupus erythematodes, revmatoidní artritida), malignity (solidní tumory, lymfoproliferace), infekce (virová hepatitida B a C) a léčiva (penicilin, interferon). Přibližně v polovině případů se nepodaří vyvolávající příčinu odhalit [1]. V patogenezi AHA se uplatňuje dosud plně neobjasněný mechanismus vzniku blokující autoprotilátky inhibitoru FVIII a jeho vazba na některé z funkčních míst FVIII a následné znemožnění vazby FVIII na FIX a FX. Dále zde působí interference s vazbou FVIII na von Willebrandův faktor, popř. interference s vazbou FVIII na fosfolipidy [4]. Nejčastěji se jedná o imunoglobuliny třídy IgG (podtřídy IgG4 v 98 %, IgG1 v 88 %), IgA v 37 %, vzácně IgM v 9 % [5]. Analýza studie GTH AH 01/2010 ukázala celkově špatnou prognózu pacientů s cirkulujícími anti FVIII třídy IgA v dosažení kompletní trvající remise a celkového přežití ve srovnání s pacienty s anti FVIII IgA negativními. U těchto nemocných se zřejmě jedná o jiný mechanismus autoimunitní patologie [5]. Potvrzení tohoto zjištění v následujících studiích může vést k cíleně odlišné imunosupresivní léčbě u anti FVIII IgA pozitivních pacientů a ke zlepšení jejich prognózy.
Klinický obraz
Krvácivé projevy u AHA jsou jiného charakteru než u vrozené hemofilie. Pacient bez osobní a rodinné anamnézy krvácivého onemocnění neužívající antikoagulační či antitrombotické léky přichází s rozsáhlou plošnou purpurou, s krvácením do měkkých tkání, svalů, sliznic (obr. 1), s dlouhotrvající epistaxí, hematurií, gastrointestinálním, popř. intracerebrálním krvácením. Na rozdíl od vrozené hemofilie krvácení do kloubů nebývá časté. Většina úvodních krvácení je neprovokovaná (77,4 %), méně často traumatická (8,4 %) nebo v souvislosti s operací (8,2 %) a peripartální (3,6 %) [2]. Výskyt závažného a život ohrožujícího krvácení, definovaného jako krvácení do kritické oblasti/orgánu 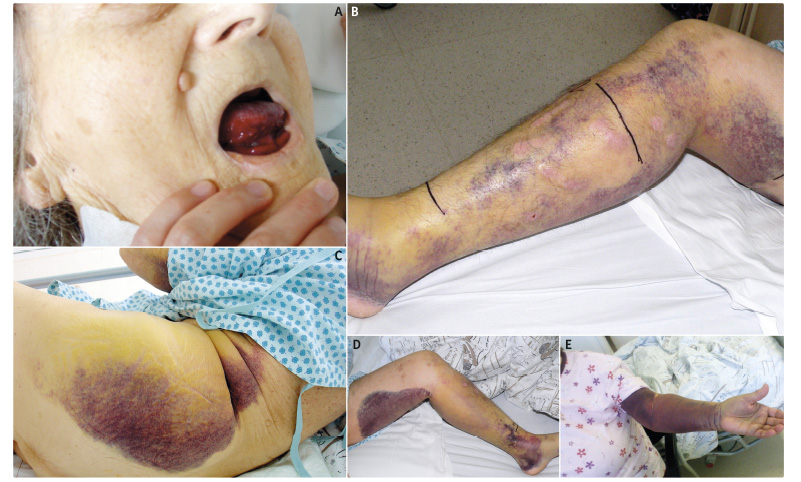 nebo krvácení s poklesem koncentrace hemoglobinu o 20 g/l a více nebo vyžadující transfuzi dvou nebo více erytrocytových transfuzních přípravků za 24 hodin nebo s poklesem koncentrace Hb pod 80 g/l, je častý (70 %) [2,3]. Se zlepšující se diagnostikou a dostupností léčby klesá úmrtnost na krvácení, ve výstupech z posledních hodnocených registrů (tab. 1) se pohybovala mezi 2,9 % až 4,5 % [1,2,6,7]. Část pacientů (6‒33 %) dokonce nekrvácí vůbec nebo má jen minimální příznaky a jediným významným projevem jsou patologické hodnoty laboratorních vyšetření zachycené i náhodně při jiném vyšetřování [8,9].
nebo krvácení s poklesem koncentrace hemoglobinu o 20 g/l a více nebo vyžadující transfuzi dvou nebo více erytrocytových transfuzních přípravků za 24 hodin nebo s poklesem koncentrace Hb pod 80 g/l, je častý (70 %) [2,3]. Se zlepšující se diagnostikou a dostupností léčby klesá úmrtnost na krvácení, ve výstupech z posledních hodnocených registrů (tab. 1) se pohybovala mezi 2,9 % až 4,5 % [1,2,6,7]. Část pacientů (6‒33 %) dokonce nekrvácí vůbec nebo má jen minimální příznaky a jediným významným projevem jsou patologické hodnoty laboratorních vyšetření zachycené i náhodně při jiném vyšetřování [8,9].
{kind=link}
Laboratorní nálezy
Typickým laboratorním nálezem u AHA je významné prodloužení hodnoty aktivovaného parciálního tromboplastinového testu (aPTT), resp. jeho poměru aPTT R na dvoj až trojnásobek při normálních hodnotách protrombinového testu i trombinového času. Korekčním směsným testem na bázi aPTT s dvouhodinovou inkubací při 37 °C (časová a teplotní závislost inhibitoru) lze odlišit deficit koagulačních faktorů tzv. vnitřní cesty od přítomnosti inhibitoru specifického i nespecifického (lupus antikoagulans). V případě deficitu koagulačních faktorů dojde po přidání normální směsné plazmy a po inkubaci ke korekci patologie, v případě inhibitoru se patologický výsledek neupraví.
Dalším krokem je zjištění specifity inhibitoru vyšetřením FVIII:C. Snížení hodnoty FVIII:C může být velmi výrazné (< 1 %), ale lze se setkat i s koncentracemi 1‒10 %, popř. i vyššími. Nakonec je nutné specifitu inhibitoru konfirmovat kvantitativním průkazem inhibitoru metodou Bethesda. Titr se uvádí v jednotkách Bethesda (Bethesda units, BU), přičemž 1 BU je množství inhibitoru, které během dvouhodinové inkubace při 37 °C inaktivuje 50 % nabídnutého faktoru. Inhibitor u AHA je inkompletně neutralizující s reakční kinetikou druhého řádu, proto může v případě nízkých titrů inhibitoru (< 10 BU/ml) docházet k podhodnocení a v případě vysokých titrů (> 20‒50 BU/ml) k jeho nadhodnocení. Je nutno zdůraznit, že hodnota FVIII:C a výše titru specifického inhibitoru FVIII často nekorelují se závažností a intenzitou klinických projevů [1,10]. V rámci laboratorní diferenciální diagnostiky je zvažována i možnost jiných koagulopatií, vrozených či získaných, včetně současné přítomnosti dalších typů inhibitorů, jako je lupus antikoagulans. Toto však přesahuje rozsah sdělení a bude komplexně řešeno v připravovaných Národních doporučeních pro diagnostiku a léčbu získané hemofilie.
Léčba získané hemofilie
Léčba pacienta s diagnózou AHA je velmi náročná a je doporučeno ji soustředit do center pro pacienty s krvácivým onemocněním, která s ní mají zkušenosti. V České republice se jedná o tzv. hemofilická centra (HTC) nebo hemofilická centra komplexní péče (HCCC). Strategie léčby AHA je směrována do tří základních kroků ‒ zástava krvácení, eradikace inhibitoru, a pokud lze, i léčba případného vyvolávajícího onemocnění. V péči o pacienta je třeba minimalizovat invazivní a diagnostické výkony jen na nezbytně nutné a o rizicích je nutné poučit také střední zdravotnický personál. I běžné výkony, jako je měření tlaku, žilní punkce, zavedení kanyly, močového katétru a podobně, mohou být vyvolávajícím momentem obtížně kontrolovatelného krvácení. Intramuskulární injekce jsou u pacientů s AHA kontraindikovány.
Léčba krvácení
U méně významných krvácení, zejména nebolestivých subkutánních, lze postupovat pouze konzervativně. U krvácejících pacientů s nízkým titrem inhibitoru (< 5 BU/ml) mohou být účinné vysoké dávky FVIII [1,14], ale vzhledem k nepredikovatelnosti hemostatického účinku (účinnost v 68 % případů dle registru EACH2) a nutnosti laboratorních kontrol FVIII:C (opakovaných rizikových venepunkcí) se jedná o alternativu pouze při nedostupnosti účinnější léčby. V současné době jsou v první linii léčby doporučovány přípravky s bypassovou aktivitou – aktivovaný rekombinantní faktor VII (eptacogum alfa, rFVIIa, NovoSeven, Novo Nordisk A/S, Dánsko) v doporučené dávce 90 µg/kg hmotnosti pacienta à 2‒3 hodiny nebo koncentrát aktivovaného protrombinového komplexu (aPCC, FEIBA NF, Baxalta Inc., USA) v dávce 50‒100 IU/kg hmotnosti pacienta à 6‒12 hodin s maximální dávkou 200 IU/kg/den [9,13]. Oba přípravky se podávají opakovaně do zástavy krvácení. Odpověď na hemostatickou terapii je sledována zejména klinicky ‒ zástava zevního krvácení, ústup bolesti při krvácení do měkkých tkání. Neexistuje specifický laboratorní test na monitoraci bypassové léčby. Účinnost obou přípravků je vysoká a srovnatelná (kolem 90 %) [14]. Pokud se nedaří zvládnout krvácení jedním z nich, je možná jejich vzájemná výměna, v případě ohrožení života u krvácení nereagujícího na monoterapii i jejich kombinace [8,9].
U slizničního krvácení, s výjimkou hematurie z horních močových cest (riziko obstrukce), je výhodné podpořit stabilitu koagula kombinací bypassové léčby s antifibrinolytiky, nejčastěji s kyselinou tranexamovou (EXACYL, sanofi aventis, Francie) v dávce 20‒25 mg 3× denně p.o., lze i ve formě 10% roztoku k výplachu dutiny ústní 1 g à 6 hodin, v nutných případech aplikace i.v. 10‒15 mg/kg 3× denně. Při renálním selhání je dávka redukována. Zvýšené opatrnosti je zapotřebí v případě kombinované léčby aPCC a antifibrinolytikem. I když jsou publikovány případy úspěšného a nekomplikovaného současného podání aPCC a kyseliny tranexamové ve dvou centrech [15], dle Souhrnu údajů o přípravku je mezi použitím obou léků doporučen minimálně šestihodinový odstup.
Léčebné možnosti nově doplňuje rekombinantní porcinní FVIII bez B domény (rFVIIIp – OBIZUR, Baxalta Inc, USA). Zkušenosti s tímto přípravkem mimo klinické studie zatím nejsou velké. Léčba rFVIIIp se zahajuje dávkou 200 IU/kg, je doporučena kontrola FVIII:C a klinického stavu 30 minut po první injekci a tři hodiny po aplikaci, dále monitorace FVIII:C před následujícími dávkami a 30 minut po nich. Dávkování a frekvence podání se řídí klinickou odpovědí a naměřenou aktivitou FVIII. Maximální hodnota FVIII:C v krvi nesmí přesáhnout 200 %. U některých pacientů mohou autoprotilátky vzniklé proti faktoru humánnímu zkříženě reagovat i s tímto faktorem porcinním, ne vždy ale negativně ovlivní jeho účinnost. U pacientů s vysokým titrem inhibitoru a život či končetinu ohrožujícím krvácením (krvácení do CNS, GIT, kompartment syndrom) či před chirurgickým výkonem je v případě dostupnosti indikována imunoadsorpce (IA). Ošetřením 2,5‒3,0 plazmatického objemu pacienta každých 24 hodin vede IA během 24‒48 hodin k rychlé kontrole krvácení, výraznému poklesu koncentrace inhibitoru a ke zkrácení doby imunosupresivní terapie. Náklady na adsorpční kolony mohou být velmi rychle kompenzovány redukovaným počtem dnů, kdy je nutno pacienta léčit bypassovými přípravky [16,17].
Eradikace inhibitoru
Ačkoliv jsou popsány případy spontánního vymizení inhibitoru, riziko závažného krvácení trvá po celou dobu jeho přítomnosti. Zahájení eradikace inhibitoru je proto doporučeno vždy a co nejdříve po stanovení diagnózy, a to i u pacientů, u nichž se krvácivé komplikace zatím neobjevily [1,13]. Imunosupresivní terapie může být spojena se závažnými nežádoucími účinky a při volbě léčebného režimu je nutno zohlednit věk (fertilita mladých žen, křehký starší polymorbidní pacient), celkový zdravotní stav, přidružená onemocnění včetně pravděpodobné primární diagnózy a další okolnosti. Při dostupnosti účinné hemostatické léčby jsou dnes častější příčinou úmrtí pacienta komplikující infekce než krvácení (tab. 1).
Optimální imunosupresivní režim k eradikaci inhibitoru v současné době není znám. V první linii léčby jsou nejčastěji používány kortikoidy – prednison v dávce 1 mg/kg hmotnosti pacienta na den samostatně nebo v kombinaci s cyklofosfamidem 1‒2 mg/kg hmotnosti pacienta na den maximálně po dobu šesti týdnů [8,9,13]. V registru EACH2 bylo dosaženo stabilní kompletní remise, definované jako nedetekovatelná přítomnost inhibitoru a FVIII:C v hodnotě nad 20 IU/dl (odpovídá 70 %) a vysazení imunosupresivní léčby, při monoterapii kortikoidy u 48 % pacientů a v případě kombinované léčby kortikoidy s cyklofosfamidem u 70 % [18], v registru UKHCDO (United Kingdom Haemophilia Center Doctors’ Organisation) byl rozdíl méně signifikantní (60‒70 % vs. 70‒80 %) [1]. Medián doby dosažení remise byl při použití tohoto způsobu léčby přibližně pět týdnů [1,18]. Pokud po 4‒6 týdnech imunosuprese nedochází k odpovědi, je doporučeno zvážit druhou linii léčby. V druhé linii je často používán rituximab (v dávce 375 mg/m² tělesného povrchu pacienta, čtyři dávky v týdenních intervalech) samostatně nebo v kombinaci s kortikoidy [9,13]. Jeho účinek v monoterapii ale nastupuje později. U pacientů v registru EACH2 byla doba potřebná k dosažení remise přibližně dvojnásobná než u pacientů léčených kortikoidy a cyklofosfamidem [18] a pro jeho přednostní indikaci u pacientů s vysokým titrem inhibitoru nemáme dostatečné důkazy [18,19]. Rituximab může být zvážen i jako lék první volby v případech, kdy jsou kontraindikována imunosupresiva (např. u postpartální AHA). Alternativou druhé linie jsou i další cytotoxické léky – cyklosporin, takrolimus, vinkristin, azathioprin, mykofenolát, popř. další. Jejich úspěšnost je přibližně 60 %, bez ohledu na použitou první linii léčby [18]. Pacienti s nižším titrem inhibitoru FVIII (méně než 16 BU/ml) a vyšší FVIII:C mají větší pravděpodobnost dosažení stabilní remise [6,18]. Část pacientů (15‒24 %) po dosažení kompletní remise (CR) relabuje v mediánu 3‒4 měsíce, proto je nezbytně nutné pravidelné klinické a laboratorní monitorování minimálně 6‒12 měsíců po dosažení CR. U režimů založených na rituximabu se zdá relaps AHA méně častý [1,18]. Ženu s anamnézou postpartální AHA je nutno informovat o riziku možného relapsu v další graviditě a každou následující graviditu je doporučeno pečlivě sledovat i hematologem specializovaným na hemostázu.
Závěr
Získaná hemofilie A je závažné onemocnění, které vyžaduje přesnou a rychlou diagnózu. Krvácení a prodloužený test aPTT u pacienta bez antikoagulační léčby a bez předchozí krvácivé anamnézy jsou prvními indiciemi vedoucími k podezření na toto onemocnění. V době dostupné účinné bypassové terapie hraje hlavní roli včasnost jejího podání a volba vhodného imunosupresivního režimu s ohledem na vyvážení rizika fatálního krvácení a častých závažných, zejména infekčních nežádoucích komplikací. Léčba pacienta s diagnózou AHA je velmi náročná a je doporučeno ji soustředit do hemofilických center.
Seznam použité literatury
- [1] llins PW, Hirsch S, Baglin TP, et al. Acquired hemophilia A in the United Kingdom: a 2‑year national surveillance study by the United Kingdom Haemophilia Centre Doctors’ Organisation. Blood 2007; 109: 1870–1877.
- [2] Knoebl P, Marco P, Baudo F, et al. Demographic and clinical data in acquired hemophilia A: results from the European Acquired Haemophilia Registry (EACH2). J Thromb Haemost 2012; 10: 622–631.
- [3] Tengborn L, Baudo F, Huth‑Kühne A, et al. Pregnancy‑associated acquired haemophilia A: results from the European Acquired Haemophilia (EACH2) registry. BJOG 2012; 119: 1529–1537.
- [4] Buliková A, Smejkal P, Zavřelová J, et al. Získané inhibitory krevního srážení. Interní Med 2008; 10: 336–339.
- [5] Tiede A, Hofbauer CHJ, Werwitzke S, et al. Anti‑factor VIII IgA as a potential marker of poor prognosis in acquired hemophilia A: results from the GTH‑AH 01/2010 study. Blood 2016; 127: 2289–2297.
- [6] Tiede A, Klamroth R, Scharf RE, et al. Prognostic factors for remission of and survival in acquired hemophilia A (AHA): results from the GTH‑AH 01/2010 study. Blood 2015; 125: 1091–1097.
- [7] Borg JY, Guillet B, Le Cam‑Duchez V, et al. Outcome of acquired haemophilia in France: the prospective SACHA (Surveillance des Auto antiCorps au cours de l’Hémophilie Acquise) registry. Haemophilia 2013; 19: 564–570.
- [8] Sborov DW, Rodgers GM. Acquired hemophilia A: a curent review of autoantibody diaease. Clin Adv Hematol Oncol 2012; 10: 19–27.
- [9] Collins P, Baudo F, Huth‑Kuhne A, et al. Consensus recommendations for the diagnosis and treatment of acquired hemophilia A. BMC Research Notes 2010; 3: 161. doi: 10.1186/1756‑0500‑3‑161
- [10] Smejkal P, Buliková A, Chlupová G, et al. Získaná hemofilie A. Vnitř Lék 2012; 58: 571–578.
- [11] Franchini M, Gandini G, Di Paolantonio T, Mariani G. Acquired hemophilia A: a concise review. Am J Hematol 2005; 80: 55–63.
- [12] Ma AD, Carrizosa D. Acquired factor VIII inhibitors: pathophysiology and treatment. Hematology Am Soc Hematol Educ Program 2006: 432–437.
- [13] Huth‑Kuhne A, Baudo F, Collins P, et al. International recommendations on the diagnosis and treatment of patients with acquired hemophilia A. Haematologica 2009; 94: 566–575.
- [14] Baudo F, Collins P, Huth‑Kühne A, et al. Management of bleeding in acquired hemophilia A: results from the European Acquired Haemophilia (EACH2) registry. Blood 2012; 120: 39–46.
- [15] Holmström M, Tran HT, Holme PA. Combined treatment with APCC (FEIBA®) and tranexamic acid in patients with haemophilia A with inhibitors and in patients with acquired haemophilia A – a two‑centre experience. Haemophilia 2012; 18: 544–549.
- [16] Zeitler H, Ulrich‑Merzenich G, Panek D, et al. Immunoadsorption in the treatment of acquired haemophilia. Atheroscler Suppl 2009; 10: 122–125.
- [17] Freedman J, Rand ML, Russell O, et al. Immunoadsorption may provide a cost‑effective approach to management of patients with inhibitors to FVIII. Transfusion 2003; 43: 1508–1513.
- [18] Collins P, Baudo F, Knoebl P, et al. Immunosuppression for acquired hemophilia A: results from the European Acquired Haemophilia registry (EACH2). Blood 2012; 120: 47–55.
- [19] Collins P, Chalmers E, Hart D, et al. Diagnosis and management of acquired coagulation inhibitors: a guideline form UKHCDO. Br J Haematol 2013; 162: 758–773.