Nové léky u akutního srdečního selhání aneb Další zklamání
Key words: acute heart failure, natriuretic peptides, vasopressin, serelaxin, positive inotropic substances.
Srdeční selhání je klinický syndrom, kdy pro nedostatečnou práci srdce dochází nejen ke zhoršení funkce řady orgánů, ale také k aktivaci mnoha humorálních působků a adaptačních mechanismů. Aktivuje se sympatikus, renin angiotenzin aldosteronový systém, vazopresin, endoteliny a cytokinový systém, které působí vesměs natrium retenčně, vazokonstrikčně a proliferativně. Na druhé straně se aktivuje systém natriuretických peptidů působící kontraregulačně. Ve farmakologii srdečního selhání se uplatňují lékové skupiny, jež potlačují tyto negativně působící mechanismy: inhibitory angiotenzin konvertujícího enzymu (ACE), blokátory receptoru AT1 pro angiotenzin II, betablokátory či blokátory aldosteronu.
U akutního srdečního selhání se nabízejí následující možnosti:
- Posílení vazodilatačních mechanismů, a to přímým podáváním natriuretických peptidů či jiných látek působících vazodilatačně, jakou je například serelaxin.
- Jsou hledány rovněž látky, které by zefektivnily diurézu (blokátory receptorů pro vazopresin či blokátory adenosinových receptorů).
- Posílení stažlivosti srdečního svalu, tedy podání inotropních látek. Výzkum se soustředí na ty látky, které nezvyšují hodnoty či tok vápníku do buněk, ale využívají vlastního vápníku prostřednictvím zvýšené citlivosti k němu (levosimendan) nebo umocňují stažlivost jiným způsobem, např. posílením vazby adenosintrifosfátu (ATP) k myosinu (omecamtiv mecarbil).
Tabulka 1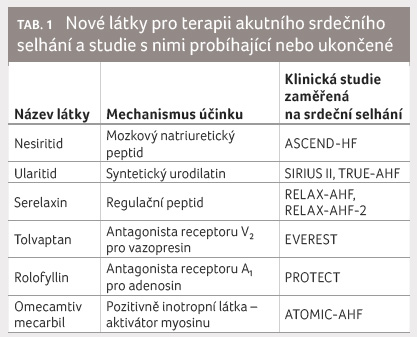 ukazuje nedávno ukončené či probíhající studie
s novými molekulami.
ukazuje nedávno ukončené či probíhající studie
s novými molekulami.
Nesiritid ve studii ASCEND HF
Studie ASCEND HF (Acute Study of Clinical Effectiveness of Nesiritide in Decompensated Heart Failure) zařadila do sledování 7 007 nemocných s akutním srdečním selháním, kteří museli být randomizováni do 24 hodin od vzniku příznaků a u nichž byl ke standardní léčbě přidán nesiritid nebo placebo na dobu 24‒168 hodin. Primárním cílovým ukazatelem bylo posouzení dušnosti na sedmistupňové škále za šest a 24 hodin a třicetidenní mortalita a rehospitalizace pro srdeční selhání. Z pohledu primárního ukazatele změny dušnosti bylo dosaženo pozitivního výsledku, protože statisticky významně více nemocných dosáhlo zlepšení stavu při podávání nesiritidu než při podávání placeba. Ukazatel třicetidenní mortality a rehospitalizace nedosáhl statistické významnosti. Studie nepotvrdila negativní vliv nesiritidu na renální funkce, látka byla z tohoto pohledu bezpečná dokonce i u pacientů s úvodně sníženou hodnotou glomerulární filtrace. Avšak výskyt hypotenze ‒ jak symptomatické, tak asymptomatické ‒ byl po podávání nesiritidu téměř dvojnásobný. Z dalších závažných nežádoucích příhod je třeba zmínit dvojnásobný výskyt reinfarktu při léčbě nesiritidem (14 vs. 6), který ale nedosáhl statistické významnosti [1].
Ularitid ve studiích SIRIUS I a II a TRUE AHF
Ularitid je syntetická forma urodilatinu, hormonu, který je produkován v buňkách distálního ledvinného tubulu. Patří mezi natriuretické peptidy. Reguluje vaskulární a renální homeostázu, zvyšuje vazodilataci a exkreci sodíku, chloridu a snižuje neurohumorální vazokonstrikční aktivaci. U akutního srdečního selhání je vyvíjena snaha o hemodynamickou stabilizaci pacienta a odstranění symptomů. Terapeutické možnosti zahrnují diuretika, vazodilatační látky a pozitivně inotropní agens. Data z metaanalýz zaměřených na používání nesiritidu poukázala na zvýšené riziko zhoršení renálních funkcí. Proto je zájmem najít látku, která by zlepšila hemodynamické parametry a zachovala renální funkce. Ve studiích SIRIUS I a II, v nichž byl ularitid podáván v případě akutní dekompenzace srdečního selhání, došlo po aplikaci 24hodinové infuze ke snížení tlaku v zaklínění a ke zmírnění dušnosti. Zlepšila se celková hemodynamika vlivem snížení vaskulární rezistence a zvýšení hodnoty srdečního indexu bez vzestupu tepové frekvence. Nežádoucí účinky byly mírné nebo střední závažnosti ‒ nejčastěji byly zaznamenány pokles krevního tlaku (5,4 %), pocení (4,2 %) a slabost (3,0 %). U pacientů léčených ularitidem bylo zachyceno méně závažných komplikací a úmrtí než u pacientů užívajících placebo [2].
Z důvodu potvrzení účinku ularitidu vzhledem k mortalitě byla naplánována klinická studie TRUE AHF (Trial of Ularitide Efficacy and Safety in Acute Heart Failure), která byla prezentována na kongresu Americké kardiologické společnosti (AHA) v roce 2016 v New Orleansu. Studie sledovala účinnost 48hodinové kontinuální infuze ularitidu (15 ng/kg/min) versus podání placeba na klinický stav pacientů s akutně dekompenzovaným srdečním selháním. Vstupními kritérii bylo přijetí pacientů k neplánované hospitalizaci pro akutní dekompenzaci srdečního selhání definovaného jako klidová dušnost, která se zhoršovala v průběhu minulých týdnů, s RTG symptomy srdečního selhání, s hodnotou mozkových natriuretických peptidů (brain natriuretic peptide, BNP) více než 500 pg/ml nebo s hodnotou N terminálního fragmentu natriuretického peptidu typu B (NT proBNP) více než 2 000 pg/ml. V době randomizace byl požadovaný systolický krevní tlak více než 116 mm Hg a méně než 180 mm Hg. Klidová dušnost měla přetrvávat navzdory terapii srdečního selhání, která musela obsahovat intravenózní dávku furosemidu 40 mg nebo více.
Primárním cílovým ukazatelem bylo zlepšení klinického stavu sestávající z hodnocení klinického stavu pacientem na sedmistupňové škále, z perzistujícího zhoršení srdečního selhání vyžadujícího intervenci (zahájení nebo intenzifikace intravenózní terapie, mechanická ventilační nebo oběhová podpora, chirurgická intervence, ultrafiltrace, hemofiltrace nebo dialýza) a z úmrtí během prvních 48 hodin. Hodnocení klinického složeného ukazatele probíhalo v 6., 24. a 48. hodině po začátku infuze. Dalším primárním cílovým ukazatelem byla kardiovaskulární mortalita sledovaná po dobu 90 dnů. K sekundárním cílovým ukazatelům patřily změna hodnoty NT proBNP za 48 hodin od počátku léčby ve srovnání se začátkem studie, délka hospitalizace, počet epizod zhoršení srdečního selhání vyžadujících intervenci v prvních 120 hodinách, změna hodnoty kreatininu v prvních 72 hodinách, riziko rehospitalizace do 30 dnů od propuštění a celková mortalita nebo kardiovaskulární rehospitalizace za 180 dnů.
K podávání ularitidu bylo
randomizováno 1 088 pacientů, 1 069 osob dostávalo
standardní léčbu a placebo. Vstupní charakteristiku souboru
ukazuje tabulka 2.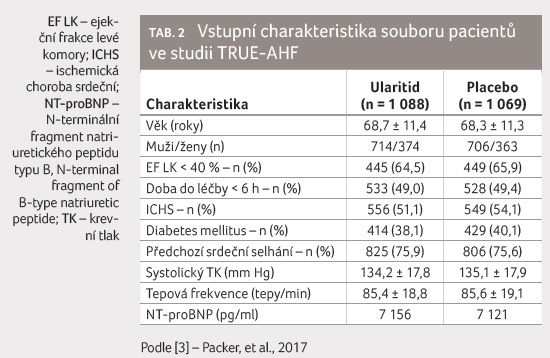
Ularitid statisticky významněji snížil krevní tlak a hodnotu NT proBNP po 48 hodinách. Došlo rovněž k významnému snížení počtu intervencí pro zhoršení nebo nelepšení se srdečního selhání po 48 hodinách (55 v případě ularitidu vs. 87 u placeba). V rámci studie však nebyl zaznamenán pokles kardiovaskulární mortality (236 úmrtí u ularitidu vs. 225 úmrtí u placeba; p = 0,75). Nedošlo k žádnému ovlivnění dalších cílových ukazatelů ‒ délky pobytu v nemocnici, epizody zhoršení srdečního selhání v průběhu 120 hodin od přijetí, rehospitalizace do 30 dnů od propuštění či kombinace celkové mortality a kardiovaskulární rehospitalizace. Z nežádoucích účinků se vyskytlo významně více hypotenze u pacientů léčených ularitidem.
Podávání ularitidu u pacientů s akutním srdečním selháním tak nenavázalo na slibné začátky z dřívějších hemodynamických studií a nepotvrdilo naděje vkládané do této nové látky [3].
Serelaxin ve studiích RELAX AHF a RELAX AHF 2
Serelaxin je lidský rekombinantní relaxin 2, původně peptid regulující adaptaci mateřského organismu na těhotenství. Má několik vlastností, které jsou využitelné v léčbě akutního srdečního selhání – zvyšuje arteriální compliance, srdeční výdej a renální průtok.
Velkou nadějí v oblasti léčby
akutního srdečního selhání byla studie RELAX AHF sledující
podávání serelaxinu oproti placebu. Tato mezinárodní, dvojitě
zaslepená randomizovaná studie zahrnula celkem 1 161 pacientů
s akutním srdečním selháním. Polovina pacientů dostávala
ke standardní terapii intravenózně po dobu 48 hodin
serelaxin, polovina pacientů placebo. Primárním cílovým
ukazatelem bylo odstranění dušnosti podle vizuální analogové
škály a podle Likertovy škály od počátku do pátého
dne sledování. Sekundárními cílovými ukazateli byly mortalita
a hospitalizace do 60. dne od počátku studie
a kardiovaskulární úmrtí nebo rehospitalizace pro srdeční
nebo renální selhání do 60. dne od zahájení
léčby. Serelaxin zmírnil dušnost pacientů podle vizuální
analogové škály (p < 0,007),
avšak ne podle Likertovy škály (p < 0,7).
Užívání látky nemělo efekt na sekundární cílové
ukazatele do 30. dne, ale překvapivě zlepšilo
dlouhodobější prognózu pacientů do 180. dne, a to
z hlediska jak kardiovaskulární, tak celkové mortality (graf 1) [4].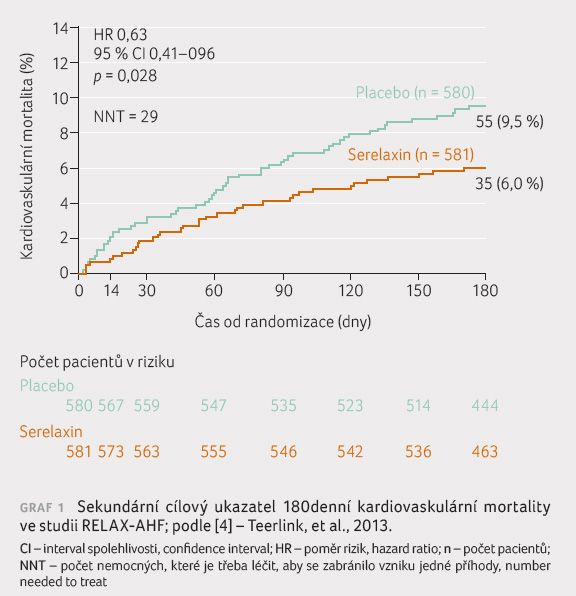
Na základě těchto pozitivních a slibných výsledků byla naplánována mortalitní studie s využitím serelaxinu a jeho srovnání s placebem u pacientů s akutně dekompenzovaným srdečním selháním RELAX AHF 2. Primárním cílovým ukazatelem byla kardiovaskulární mortalita za 180 dnů a zhoršení srdečního selhání do pěti dnů od přijetí. Sekundárním složeným cílovým ukazatelem byla 180denní celková mortalita, délka hospitalizace a kardiovaskulární úmrtí nebo rehospitalizace pro srdeční či renální selhání do 180 dnů.
Do studie byli zahrnuti pacienti, kteří byli hospitalizováni pro akutní srdeční selhání. Klinicky měli známky dušnosti, městnání na RTG snímku, zvýšené hodnoty BNP více než 500 pg/ml nebo NT proBNP více než 2 000 pg/ml. U nemocných ve věku nad 75 let nebo s fibrilací síní byl požadavek na vyšší vstupní hodnoty BNP a NT proBNP ‒ více než 750 pg/ml, resp. více než 3 000 pg/ml ‒, dále systolický krevní tlak více než 125 mm Hg a mírné renální poškození (glomerulární filtrace více než 25 ml/min/1,73 m2 a méně než 75 ml/min/1,73 m2). Pacienti měli být symptomatičtí i po podání dávky 40 mg furosemidu intravenózně. Ejekční frakce levé komory nebyla kritériem pro zařazení, takže ve studii mohli být nemocní jak se sníženou, tak se zachovanou ejekční frakcí levé komory. Pacienti byli randomizováni do 16 hodin od obdržení první dávky diuretika nebo od příjezdu do nemocnice. V průběhu hospitalizace byli nemocní do pátého dne denně klinicky sledováni, následně pak v den 14, 60, 120 a 180 [5].
Výsledky studie byly poprvé prezentovány profesorem Teerlinkem a profesorem Metrou v sekci Hot lines na evropském kongresu Heart Failure v dubnu 2017 v Paříži [6,7]. Celkem bylo screenováno 7 554 pacientů ve 35 zemích, randomizováno bylo 3 274 osob k užívání serelaxinu a 3 271 osob k užívání placeba. Předchozí srdeční selhání měly tři čtvrtiny nemocných, nejčastější diagnózou vedoucí k srdečnímu selhání byla ischemická choroba srdeční, a to ve více než polovině případů. Z komorbidit byla nejčastější hypertenze (90 %) a hyperlipoproteinemie (50 %), následovaná diabetes mellitus ve 46 % případů.
Primární cílový ukazatel kardiovaskulární mortality za 180 dnů se vyskytl u 8,7 % pacientů v léčené skupině a u 8,9 % pacientů v placebové větvi (statisticky nesignifikantně, NS) [6,7]. Primární parametr zhoršení srdečního selhání do pátého dne se objevil u 6,9 % osob léčených serelaxinem a u 7,7 % osob v placebové větvi (NS) [6,7]. Z dalších hodnocených sekundárních cílových ukazatelů nebyly nalezeny rozdíly v celkové mortalitě za 180 dnů (u serelaxinu 11,2 % a u placeba 11,9 %). Rovněž sekundární složený cílový ukazatel kardiovaskulárního úmrtí nebo rehospitalizace pro srdeční selhání se nelišil (u serelaxinu 24,3 % vs. 24,9 % u placeba; NS) [6,7].
Ačkoliv dřívější studie se serelaxinem prokázaly hemodynamickou účinnost a naději, že dojde i ke zlepšení přežívání pacientů se srdečním selháním, studie RELAX AHF 2 nepotvrdila účinek této látky v případě kardiovaskulární mortality po 180 dnech či zlepšení srdečního selhání do pátého dne.
Tolvaptan ve studii EVEREST
U srdečního selhání dochází k objemovému přetížení na základě retence tekutin, které je způsobeno zvýšením koncentrací arginin vazopresinu. Tolvaptan je blokátor receptoru V2 pro vazopresin a tímto způsobem antagonizuje jeho účinek. EVEREST byla randomizovaná multicentrická, mezinárodní dvojitě zaslepená studie kontrolovaná placebem, která sledovala účinek tolvaptanu u pacientů s dekompenzací srdečního selhání. Celkem do ní bylo zařazeno 4 133 osob. Během sledování (medián 9,9 měsíce) zemřelo 537 (25,9 %) pacientů léčených tolvaptanem a 543 (26,3 %) pacientů v placebové větvi (p = 0,68). Složený cílový ukazatel kardiovaskulární mortality a hospitalizací pro srdeční selhání se rovněž nelišil (p = 0,55). Podání tolvaptanu vedlo ke zmírnění dušnosti hodnocené nemocnými v prvním dnu a ke snížení hmotnosti první den. V laboratorním sledování bylo zaznamenáno zvýšení koncentrace sodíku. Z nežádoucích účinků se u pacientů léčených tolvaptanem častěji vyskytla suchost v ústech a žízeň. Lze tedy říci, že použití tolvaptanu u nemocných s akutní dekompenzací srdečního selhání vede při akutním podání ke klinické úlevě, avšak při dlouhodobějším sledování nedochází ke zlepšení prognózy nemocných [8].
Rolofyllin ve studii PROTECT
Rolofyllin je látka, která blokuje receptor A1 pro adenosin. Stimulací receptoru A1 pro adenosin na aferentní arteriole glomerulu snižuje adenosin renální průtok a glomerulární filtraci a v proximálním tubulu zvyšuje reabsorpci sodíku a vody. Rolofyllin způsobuje vzestup diurézy, glomerulární filtrace a renálního průtoku u pacientů se srdečním selháním.
Studie PROTECT sledovala účinek rolofyllinu na renální funkce u nemocných s akutním srdečním selháním a s renální dysfunkcí. Do sledování bylo zařazeno 2 033 pacientů s akutním srdečním selháním, s objemovým přetížením, s clearance kreatininu mezi 20‒80 ml/min a se zvýšenou koncentrací natriuretických peptidů. Změny v hodnotách kreatininu a clearance kreatininu se u placebové skupiny a skupiny léčené rolofyllinem v průběhu studie nelišily ani ve 14. dnu. Perzistentní zhoršování renálních funkcí se vyskytlo u 13,7 % osob v placebové skupině a u 15 % osob ve skupině léčené rolofyllinem. Tato studie přinesla zklamání, protože nepotvrdila protektivní účinek rolofyllinu na renální funkce u pacientů přijatých do nemocnice s akutním srdečním selháním [9]. Při hodnocení prognózy nemocných byla sledována třicetidenní mortalita nebo rehospitalizace z jakékoliv příčiny, třicetidenní mortalita nebo rehospitalizace z důvodů kardiovaskulárních či renálních a třicetidenní nebo 180denní celková mortalita. Při podávání rolofyllinu nevyšel žádný z těchto parametrů statisticky významně lépe oproti placebu a oproti standardní léčbě srdečního selhání [10].
Omecamtiv mecarbil ve studii ATOMIC AHF
Pozitivně inotropní látky původně zvyšovaly kontraktilitu levé komory vzestupem cyklického adenosinmonofosfátu a tím rovněž cytosolového vápníku (sympatomimetika či inhibitory fosfodiesterázy). Akutní krátkodobé podání těchto léků může být pro vybrané pacienty prospěšné, obecně je zvyšování hodnoty cytosolového vápníku u pacientů se srdečním selháním škodlivé. I když se posílí systolická funkce levé komory, děje se tak na úkor zvýšené spotřeby kyslíku myokardem, což vede jak ke vzestupu výskytu arytmií, tak k ischemii. Novým přístupem, jímž by se posílila srdeční stažlivost bez nutnosti zvýšení hodnot nitrobuněčného vápníku, je aktivace vydatnějšího stahu prostřednictvím samotného proteinu ‒ srdečního myosinu.
Omecamtiv mecarbil je látka, která selektivně aktivuje srdeční myosin. Během kontrakce se myosin váže na aktin a tvoří můstky (cross bridges), což vede ke zkrácení vlákna. Omecamtiv mecarbil zrychluje přeměnu slabého komplexu aktin myosin na silně vázanou konfiguraci, která zvyšuje počet myosinových hlavic interaktivních s aktinovými filamenty a současně snižuje míru neproduktivní ATP hydrolýzy. Tento stav s větším množstvím myosinových hlavic na aktinovém vláknu je možné přirovnat k situaci více rukou na provaze a tím silnějšího stahu. Tento děj je nezávislý na vápníkové homeostáze a kyslíku, zlepšení funkce levé komory by tedy nemělo nastat na úkor zvýšení spotřeby energie a arytmogenicity [11].
První studie sledovala podávání látky u 34 zdravých dobrovolníků, kteří dostávali šestihodinovou infuzi omecamtivu mecarbilu nebo placebo jednou týdně po dobu čtyř týdnů. Infuze omecamtivu mecarbilu vedla k prodloužení systolické ejekční doby, jež byla spojena se zvýšením tepového objemu, s frakčním zkrácením a se zvýšením ejekční frakce levé komory. Účinek byl závislý na dávce léku. Nedocházelo ke změnám diastolické funkce [12].
Dalším krokem bylo podání omecamtivu mecarbilu pacientům se srdečním selháním. První taková studie zahrnula 45 pacientů, kteří dostali 151 infuzí aktivní látky nebo placebo. U aktivně léčených pacientů došlo k prodloužení ejekční doby levé komory a ke zvýšení srdečního tepového objemu spojeného s mírným snížením tepové frekvence. Vyšší plazmatické koncentrace sledované látky znamenaly pokles enddiastolického a endsystolického objemu [13].
Vzhledem k těmto příznivým hemodynamickým výsledkům byla zahájena klinická studie ATOMIC AHF sledující 606 pacientů s dekompenzací srdečního selhání, s ejekční frakcí levé komory méně než 40 % a se zvýšenou koncentrací BNP (> 400 pg/ml) a NT proBNP (> 1 600 pg/ml). Nemocní byli randomizováni ke třem různým dávkám omecamtivu mecarbilu a k podávání placeba. Omecamtiv mecarbil nebyl lepší než placebo v rámci primárního cílového ukazatele zmírnění dušnosti za 6, 24 a 48 hodin ani v žádném ze sekundárních cílových ukazatelů ‒ mortalita do sedmého dne, zhoršení srdečního selhání do sedmého dne, délka hospitalizace, třicetidenní přežívání a změna koncentrace natriuretických peptidů. Došlo k prodloužení ejekčního systolického času. Tolerance léku byla dobrá, nezvýšil se výskyt komorových arytmií. Laboratorně došlo u pacientů léčených omecamtivem mecarbilem ke zvýšení koncentrace troponinu bez klinického ovlivnění stavu [14].
I přes neutrální výsledek studie ATOMIC AHF byla naplánována a provedena studie COSMIC u pacientů s chronickým srdečním selháním s podáváním perorální formy této látky, která rovněž nepřinesla zásadní pozitivní výstupy.
Závěr
Oblast srdečního selhání, zejména akutní dekompenzace, je bohatá na nové lékové skupiny. Klinické studie zaměřené na symptomatologii či hemodynamiku prokazují u pacientů zlepšení v těchto parametrech. Z hlediska dlouhodobé prognózy však u žádné z těchto lékových skupin pozitivní efekt potvrzen nebyl.
Seznam použité literatury
- [1] O’Connor CM, Starling RC, Hernandez AF, et al. Effect of nesiritide in patients with acute decompensated heart failure. N Engl J Med 2011; 365: 32‒43.
- [2] Mitrovic V, Seferovic PM, Simeunovic D, et al. Haemodynamic and clinical effects of ularitide in decompensated heart failure. Eur Heart J 2006; 27: 2823‒2832.
- [3] Packer M, O’Connor C, McMurray JJV, et al. Effect of Ularitide on Cardiovascular Mortality in Acute Heart Failure. N Engl J Med 2017; 376: 1956‒1964.
- [4] Teerlink JR, Cotter G, Davison BA, et al. Serelaxin, recombinant human relaxin‑2, for treatment of acute heart failure (RELAX‑AHF): a randomised, placebo controlled trial. Lancet 2013; 381: 29‒39.
- [5] Teerlink JR, Voors AA, Ponikowski P, et al. Serelaxin in addition to standard therapy in acute heart failure: rationale and design of the RELAX‑AHF‑2 study. Eur J Heart Fail 2017; 19: 800‒809.
- [6] Teerlink JR. Late breaking trials I, Focus on heart failure. RELAX‑AHF‑2: Serelaxin in acute heart failure, Part 1. Congresses Heart Failure 2017 ‒ 4th World Congress on Acute Heart Failure, Paris. Dostupné na: http://spo.escardio.org/
- [7] Metra M. Late breaking trials I, Focus on heart failure. RELAX‑AHF‑2: Serelaxin in acute heart failure, Part 2. Congresses Heart Failure 2017 ‒ 4th World Congress on Acute Heart Failure, Paris. Dostupné na: http://spo.escardio.org/
- [8] Konstam MA, Gheorghiade M, Burnett C, et al. Effects of oral tolvaptan in patients hospitalised for worsening heart failure. JAMA 2007; 297: 1319‒1331.
- [9] Voors AA, Ditrich HC, Massie BM, et al. Effects of the adenosine A1 receptor antagonist rolofylline on renal function in patients with acute heart failure and renal dysfunction. J Am Col Cardiol 2011; 57: 1899‒1907.
- [10] Metra M, O’Connor CM, Davison BA, et al. Early dyspnoea relief in acute heart failure: prevalence, association with mortality, and effect of rolofylline in the PROTECT Study. Eur Heart J 2011; 32: 1519–1534.
- [11] Malik FI, Hartman JJ, Elias KA, et al. Cardiac Myosin Activation: A Potential Therapeutic Approach fo Systolic Heart Failure. Science 2011; 331: 1439‒1443.
- [12] Teerlink JR, Clarke CP, Sfikali KG, et al. Dose‑dependent augmentation of cardiac systolic function with the selective cardiac myosin activator, omecantiv mecarbil: a first‑in‑man study. Lancet 2011; 378: 667‒675.
- [13] Cleland JGF, Teerlink JR, Senior R, et al. The effects of the cardiac myosin activator, omecantiv mecarbil, on cardiac function in systolic heart failure: a double‑blind, placebo‑controlled, crossover, dose‑ranging phase 2 trial. Lancet 2011; 378: 676‒683.
- [14] Teerlink JR, Felker M, McMurray JJV, et al. Acute tratment with omecamtiv mecarbil to increase contractility in acute heart failure: The ATOMIC‑AHF study. J Am Coll Cardiol 2016; 67: 1444–1455.