Význam farmakogenetiky v léčbě srdečního selhání
Léčba srdečního selhání je zaměřena na tři oblasti – na úpravu hyperaktivovaných regulačních mechanismů (betablokátory, inhibitory ACE, inhibitory mineralokortikoidních receptorů a eventuálně antagonisté receptorů AT1 ), na úpravu postižené kontraktility a zlepšení perfuze tkání (zejména inotropika, event. vazodilatancia) a na snížení retence tekutin (diuretika).
Genetická výbava jistě ovlivňuje výskyt i průběh srdečního selhání, nicméně naše znalosti jsou v této oblasti ještě nedostatečné a jejich přímé využití v klinické praxi zatím není možné. Na straně druhé polymorfismy na úrovni absorpce, transformace a eliminace léčiv již jsou v základním obrysu zmapované a farmakogenetické principy již v léčbě respektujeme. Zejména biologická dostupnost a eliminace, stejně jako délka působení lipofilních betablokátorů, eventuálně digoxinu, v závislosti na farmakogenetické výbavě je významná. Odlišnosti ve farmakodynamické odpovědi léčiv v závislosti na polymorfismech cílových struktur působení léků (enzymů, transportních a iontových kanálů apod.) jsou již prozkoumány méně, avšak minimálně na úrovni betablokátorů a inhibitorů ACE probíhá intenzivní výzkum.
Je pouze málo oblastí medicíny, které prodělaly tak významný pokrok v léčbě, jako je tomu v terapii srdečního selhání. Během jedné či dvou generací se nám do rukou dostaly postupy významně zlepšující prognózu nemocných (graf 1). Zásluhu na tom má zavedení principů medicíny založené na důkazech, tedy využití poznatků z klinických intervenčních studií. Výsledky „megastudií“ platí pro populaci, ale nemusí nutně platit pro jednotlivce. Léčebná odpověď je totiž velmi individuální, lišíme se nejen v oblasti ovlivnění farmakokinetiky léčiva (dostupnost, distribuce, transformace i eliminace), ale také v míře farmakodynamické odpovědi na daný lék. Zde, vedle ovlivnění expozice, se uplatní vliv individ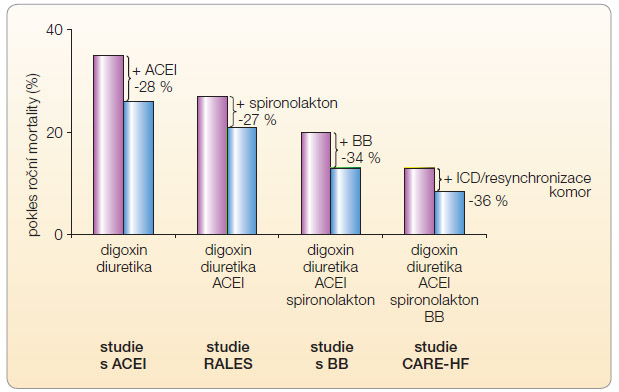 uální odpovědi organismu, tj. uplatní se citlivost řady cílových struktur k působení léčiva, tj. enzymů, receptorů, iontových kanálů a regulačních, transportních či transkripčních proteinů.
uální odpovědi organismu, tj. uplatní se citlivost řady cílových struktur k působení léčiva, tj. enzymů, receptorů, iontových kanálů a regulačních, transportních či transkripčních proteinů.
Shrneme-li, pak jak farmakokinetika, tak farmakologická odpověď je pod vlivem řady polymorfismů, je ovlivňována farmakogenetickou výbavou jednotlivce. Tyto faktory mohou vést až k desetinásobným interindividuálním rozdílům v účinku. Rozdíly v expozici mají přímý dopad na jedné straně selháním léčby, na straně druhé toxickým působením. Klinicky významně se pak uplatní zejména u léčiv s úzkým terapeutickým oknem, typickými příklady jsou antikoagulancia s rizikem trombózy či krvácení či kardiotonika nebo antiarytmika s nebezpečím navození život ohrožující dysrytmie. Vedle antitrombotické léčby je význam farmakogenetiky v léčbě srdečního selhání nejzásadnější.
Farmakogenetické faktory ovlivňující výsledný efekt léku
Farmakogenetické faktory ovlivňující osud léku v organismu (ovlivnění farmakokinetiky)
O účinku léku rozhodují nejen jeho vlastnosti farmakodynamické, ale též jeho biologická dostupnost, biotransformace a eliminace, tedy jeho vlastnosti farmakokinetické. Zatímco farmakodynamický účinek je více méně stabilní, variabilita v expresi cílových struktur účinku léku – receptorů, enzymů či iontových kanálů – nebývá veliká, rozdíly v expozici léčivu jsou často podstatné. V prvém kroku je osud léčiva ovlivněn biologickou dostupností, která je ovlivněna aktivitou transmembranózních transportérů a metabolických systémů na úrovni enterocytu i jater. Na úrovni enterocytu ovlivňují absorpci zejména eliminační transportní proteiny. Ty fungují jako 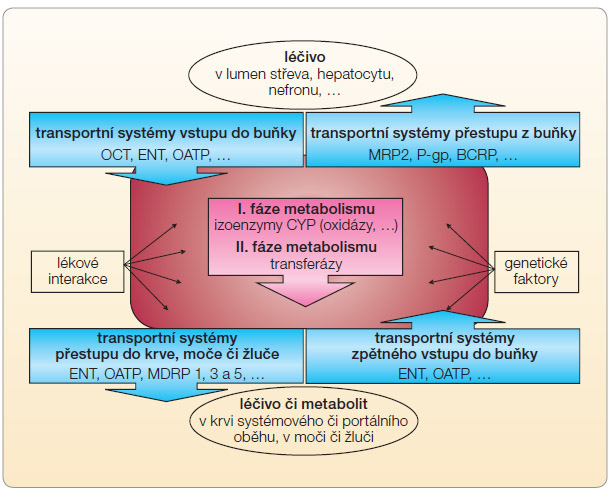 pumpy mající funkci „gate-keeperů“, které zabraňují vstřebání cizorodých látek a vylučují je z buňky zpět do lumina střeva. Obdobně jsou těmito proteiny osazeny epitelie nefronu a žlučových cest, kdy je aktivně stimulována eliminace xenobiotika do moče a žluče. Na regionální úrovni tyto transportní pumpy zajišťují funkci hematoencefalické, testikulární či placentární bariéry. Funkce těchto efluxních systémů je energeticky závislá na ATP, proto jsou označovány rodinným jménem – transportéry ABC (ATP-binding cassette), další písmeno označuje typ izoproteinu. Aktivita všech těchto pump je výrazně ovlivněna farmakogenetickou výbavou či lékovými interakcemi (obr. 1).
pumpy mající funkci „gate-keeperů“, které zabraňují vstřebání cizorodých látek a vylučují je z buňky zpět do lumina střeva. Obdobně jsou těmito proteiny osazeny epitelie nefronu a žlučových cest, kdy je aktivně stimulována eliminace xenobiotika do moče a žluče. Na regionální úrovni tyto transportní pumpy zajišťují funkci hematoencefalické, testikulární či placentární bariéry. Funkce těchto efluxních systémů je energeticky závislá na ATP, proto jsou označovány rodinným jménem – transportéry ABC (ATP-binding cassette), další písmeno označuje typ izoproteinu. Aktivita všech těchto pump je výrazně ovlivněna farmakogenetickou výbavou či lékovými interakcemi (obr. 1).
Nejvýznamnějšími transportéry jsou P-glykoprotein (P-gp, též MDR1, kódovaný genem ABCB1) a breast cancer resistance protein (BCRP, též ABCG2). Paralelně jsou oba póly buňky osazeny influxními transportními proteiny (resp. glykoproteiny), které naopak absorpci potencují. Významným příkladem je organic anion transporting polypeptid (OATP)1B1, který může ovlivnit jak vstup do buňky, tak eliminaci z buňky. Pumpa OATP je významná zejména v hepatocytu, zde facilituje na straně kapilární vstup léčiva do buňky a na straně biliární jeho eliminaci do žluče. Typickým příkladem významu absence této pumpy je léčba lipofilními statiny. Genotypy typu „loss of function“, tedy se sníženou aktivitou, vyskytující se u 15 % populace, jsou sdruženy s poruchou eliminace statinů (simva- > atorva- > rosuva- > fluvastatinu) a s vyšším výskytem myalgií a myopatií (5krát vyšší u heterozyzotů, 30krát vyšší u homozygotů). Navíc, vzhledem k nízké koncentraci statinu v hepatocytu, se zdá být hypolipidemický efekt statinu u těchto osob snížen [1, 2].
Z klinického pohledu má největší význam polymorfismus P-gp. Je popsáno téměř 30 mutací kódujícího genu ABCB1. Řada mutací, zejména v pozici 2677 či 3435, je spojena s alterací exprese P-gp, která je provázena změnou jeho aktivity, nebo se změnou specificity k substrátu. Asi u čtvrtiny naší populace, nosičů genotypu 3435TT (ABCB1 3435C→T), se setkáváme s vysokou expresí P-gp a s nižší dostupností substrátu proti nosičům alely 3435CT. Naopak u jiné čtvrtiny populace, u nosičů genotypu 3435CC, je známa nižší aktivita P-gp spojená s vysokou dostupností substrátu a větším klinickým efektem [3]. Rozdíly v hladinách substrátů P-gp se pak interindividuálně liší o více než dvojnásobek. Ze substrátů P-gp a současně léků s úzkým terapeutickým oknem je nutno zdůraznit např. digoxin, většinu antiarytmik či nová přímá antikoagulancia.
Vedle absorpce a eliminace ovlivňuje osud léčiva v organismu biotransformace, tedy jeho aktivace či inaktivace.
Řada metabolických systémů modifikuje molekuly tělu vlastních látek či látek cizorodých oxidací (aktivitou hydroláz, esteráz apod.) či vnesením hydrofilní skupiny (glukuronidací, nitrosylací apod.). Tak se zvýší rozpustnost ve vodě a usnadní se eliminace či se změní biologická aktivita – aktivací či inaktivací. Oxidace molekul je zprostředkována rodinou izoenzymů cytochromového systému P450 (CYP). Z transferáz, tedy enzymů konjugujících lipofilní molekulu s hydrofilními radikály, je nejvýznamnější glukuronyltransferáza. Většina z enzymů CYP i enzymů konjugačních je významně polymorfních.
Zejména na úrovni cytochromového systému bylo odhaleno velké množství polymorfismů. Některé vedou ke snížení aktivity enzymu až k jeho naprosté neúčinnosti, jiné naopak efekt enzymu zvyšují. Výsledný účinek závisí na charakteru substituce i na tom, zda má jedinec daným polymorfismem postiženu jednu, či obě alely, homozygotní postižení mění aktivitu výrazněji. U některých oxidáz CYP, typicky u oxidázy CYP2D6 metabolizující lipofilní betablokátory, se setkáváme s multiplikací alel a s velmi vysokou expresí izoenzymu. Je logické, že takovéto změny se odrazí v osudu léku v organismu, tj. v jeho farmakokinetice.
Pod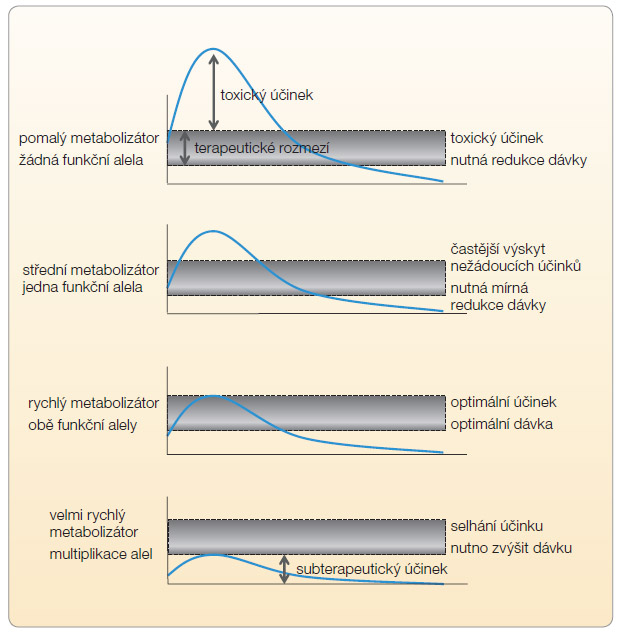 le genotypu pro daný CYP (např. 2D6 či 2C9) pak rozlišujeme populaci na pomalé, střední, rychlé a ultrarychlé metabolizátory. Běžná dávkovací schémata léků pak často vyhovují pro střední, event. rychlé metabolizátory, u pomalých dochází ke kumulaci až k toxickým hladinám léčiva, či naopak u velmi rychlých metabolizátorů hladina léčiva po většinu dávkovacího období nedosáhne terapeutických hodnot, a léčba je neúčinná (obr. 2).
le genotypu pro daný CYP (např. 2D6 či 2C9) pak rozlišujeme populaci na pomalé, střední, rychlé a ultrarychlé metabolizátory. Běžná dávkovací schémata léků pak často vyhovují pro střední, event. rychlé metabolizátory, u pomalých dochází ke kumulaci až k toxickým hladinám léčiva, či naopak u velmi rychlých metabolizátorů hladina léčiva po většinu dávkovacího období nedosáhne terapeutických hodnot, a léčba je neúčinná (obr. 2).
Rychlost degradace některých léků daným systémem se pak může podle genotypu odlišovat deseti- až tisícinásobně. Naštěstí však, díky překrývání substrátů, bývá lék často eliminován či biotransformován též jiným systémem a v praxi se interindividuální koncentrace léku odlišují o jeden, maximálně o dva řády. Příkladem je antiarytmikum propafenon, metabolizované polymorfním izoenzymem CYP2D6, kde u pomalých metabolizátorů při běžném dávkování dosahuje hladina propafenonu a jeho aktivního metabolitu kardiotoxických hodnot, zatímco u velmi rychlých metabolizátorů zůstáváme pod terapeutickou hladinou. Pro klinika je důležité, že některé typy polymorfismu na úrovni transportu či metabolismu léku se vyskytují v populaci až v desítkách procent a v praxi musíme jejich přítomnost zohlednit.
Farmakogenetické faktory ovlivňující odpověď organismu na lék (ovlivnění farmakodynamiky)
Každé léčivo působí na jeden či více cílových receptorů, enzymů, iontových kanálů nebo regulačních proteinů. Tato aktivita stimuluje či inhibuje danou funkci a zajistí farmakodynamickou odpověď (obr. 3). V současné době známe stovky polymorfismů těchto cílových struktur, kdy snížení, či naopak zvýšení odpovědi na podnět může významně ovlivnit odpověď na lék. V některých případech stačí polymorfismus jednoho receptoru k patologické reakci, jindy je nutná souhra polymorfismů dvou nebo více receptorů. Obdobně je tomu s enzymy, kanály či regulačními molekulami.
V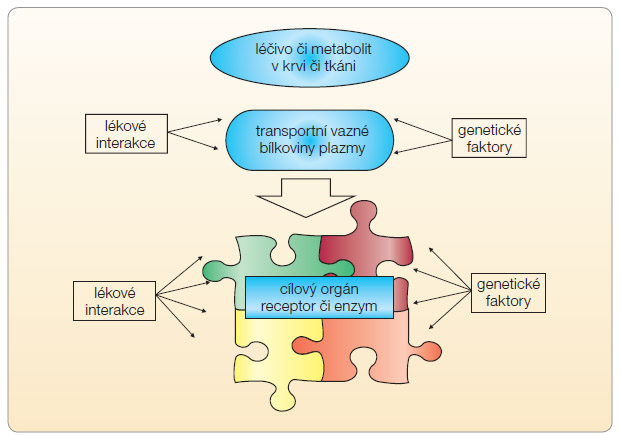 léčbě srdečního selhání se např. uplatní variabilní citlivost adrenergního receptoru β či α (ARβ, ARα) ke stimulaci a tak je ovlivněna i výsledná odpověď efektoru (např. buněk převodního systému, myokardu či hladké svaloviny arteriol). Ve většině případů nestačí k významnější modifikaci efektu systému změna aktivity jednoho receptoru, regulační mechanismy zpravidla takovýto výkyv z rovnováhy kompenzují. K patologické odpovědi bývá nutná souhra mutací na dvou i více místech, např. na dvou spolupracujících receptorech. Takovým příkladem je polymorfismus pre- a postsynaptického receptoru. Adrenergní aktivace myokardu je pod kontrolou presynaptického receptoru ARα2c, který inhibuje uvolnění noradrenalinu z nervového zakončení a z receptoru postsynaptického ARα1 zprostředkovávajícího vlastní odpověď kardiomyocytu na uvolnění noradrenalinu. Jsou známy polymorfismy presynaptického receptoru spojené s jeho sníženou inhibiční aktivitou a polymorfismus postsynaptického receptoru spojený naopak s hyperaktivitou. Izolované polymorfismy ARα2c (Del32–325) a ARα1 (arg389), které jsou v populaci přítomny asi v 10–30 %, nemění odpověď na stimulaci, hyperaktivita jednoho je kontrolována fyziologickým útlumem druhého a naopak. Teprve přítomnost obou polymorfismů na nervovém zakončení, tj. výraznější stimulační aktivita postsynaptického a nedostatečná inhibiční odpověď presynaptického receptoru, vede k α-adrenergní hyperstimulaci myokardu a ke zvýšenému efektu adrenergní blokády např. karvedilolem. Vliv jednotlivých polymorfismů adrenergních receptorů na intenzitu farmakodynamické odpovědi na stimulaci či na blokádu receptoru je dobře doložen, naopak nebyl potvrzen dopad jednotlivých polymorfismů na výskyt a průběh kardiovaskulárních chorob [4].
léčbě srdečního selhání se např. uplatní variabilní citlivost adrenergního receptoru β či α (ARβ, ARα) ke stimulaci a tak je ovlivněna i výsledná odpověď efektoru (např. buněk převodního systému, myokardu či hladké svaloviny arteriol). Ve většině případů nestačí k významnější modifikaci efektu systému změna aktivity jednoho receptoru, regulační mechanismy zpravidla takovýto výkyv z rovnováhy kompenzují. K patologické odpovědi bývá nutná souhra mutací na dvou i více místech, např. na dvou spolupracujících receptorech. Takovým příkladem je polymorfismus pre- a postsynaptického receptoru. Adrenergní aktivace myokardu je pod kontrolou presynaptického receptoru ARα2c, který inhibuje uvolnění noradrenalinu z nervového zakončení a z receptoru postsynaptického ARα1 zprostředkovávajícího vlastní odpověď kardiomyocytu na uvolnění noradrenalinu. Jsou známy polymorfismy presynaptického receptoru spojené s jeho sníženou inhibiční aktivitou a polymorfismus postsynaptického receptoru spojený naopak s hyperaktivitou. Izolované polymorfismy ARα2c (Del32–325) a ARα1 (arg389), které jsou v populaci přítomny asi v 10–30 %, nemění odpověď na stimulaci, hyperaktivita jednoho je kontrolována fyziologickým útlumem druhého a naopak. Teprve přítomnost obou polymorfismů na nervovém zakončení, tj. výraznější stimulační aktivita postsynaptického a nedostatečná inhibiční odpověď presynaptického receptoru, vede k α-adrenergní hyperstimulaci myokardu a ke zvýšenému efektu adrenergní blokády např. karvedilolem. Vliv jednotlivých polymorfismů adrenergních receptorů na intenzitu farmakodynamické odpovědi na stimulaci či na blokádu receptoru je dobře doložen, naopak nebyl potvrzen dopad jednotlivých polymorfismů na výskyt a průběh kardiovaskulárních chorob [4].
Druhým mechanismem, který se uplatní v léčbě srdečního selhání, je ovlivnění regulační funkce adrenergní stimulace. Intenzita sympatoadrenální aktivity reguluje expresi adrenergních receptorů β nebo α na buněčné membráně, hyperstimulace vede k down-regulaci, tedy ke snížení koncentrace, a naopak nízká stimulace vede k up-regulaci receptoru. Zejména u srdečního selhání je tato regulační funkce velmi významná, chrání myokard před toxickým vlivem chronické adrenergní hyperstimulace. V populaci velmi častá (desítky procent) varianta ARβ typu Gly49 je ve srovnání s běžnou variantou Ser49 spojena s výraznější odpovědí (down-regulací) na stimulaci adrenergním agonistou, tedy je přítomen protektivní prvek [5]. Paralelně je doložena výraznější odpověď této genetické varianty na inhibici adrenergní aktivity metoprololem v experimentu [6]. Význam genotypu pro průběh srdečního selhání či vliv na remodelaci levé komory je testován, dosavadní nálezy zatím nedovolují jednoznačnou interpretaci.
U komplikovanějších regulačních systémů bývá situace obvykle ještě složitější, tak například u systému renin-angiotenzin-aldosteron (RAAS) se uplatní polymorfismy na úrovni angiotenzinogenu, angiotenzin konvertujícího enzymu (ACE), receptoru AT1, (pro)reninového receptoru, bradykininového receptoru či receptoru MAS pro angiotenzin 1–7. Výsledná mozaika odchylek pak určuje aktivitu systému i farmakoterapeutickou odpověď. Kvůli komplikovanosti je tato oblast méně objasněna a jen vzácně lze přímo přenést farmakogenetickou odchylku do reálné praxe.
Jak je patrné z předložených dat, význam farmakogenetiky je široký, interindividuálními rozdíly se projeví ve vstřebávání, distribuci, eliminaci či aktivaci léku, respektive ovlivní odpověď cílové struktury. Respektování interindividuálních odlišností v léčbě se prosazuje jen pomalu. Příčin je více, zejména se uplatní minimální zkušenosti kliniků s farmakogenetickým testováním, neznámý terén pak vede ke zdrženlivosti. Pravdou je i skutečnost, že interpretace výsledků není jednoduchá a často máme v rovnici příliš mnoho neznámých. Nicméně není pochyb, že dříve nebo později musíme realizovat posun od strategie „wait and see“ ke strategii „measure and manage“.
Význam farmakogenetiky v základních strategiích farmakoterapie srdečního selhání
Léčebná strategie chronického srdečního selhání je cílena na tři oblasti – potlačení hyperaktivovaných regulačních mechanismů (konkrétně sympatoadrenální aktivace a RAAS), zlepšení funkce levé komory (pozitivně inotropními léky, snížením diastolické náplně levé komory při její dilataci, snížením periferní rezistence, eventuálně postupy redukujícími případnou myokardiální ischemii) a snížení retence tekutin (v plicní a systémové cirkulaci). Při ovlivnění prognózy hraje hlavní úlohu právě inhibice hyperaktivované sympatické regulace betablokátory a blokáda účinku angiotenzinu II (zejména inhibitory ACE) a aldosteronu (inhibitory mineralokortikoidních receptorů). Inotropika (digoxin, sympatomimetika či levosimendan) mají význam při řešení akutního stavu, digoxin dlouhodobě mírně zlepší kvalitu života, ovlivnění prognózy doloženo není. Obdobně diuretika zlepšují zásadním způsobem kvalitu života, prognostická studie však chybí. Efekt uvedených léčebných postupů je často ovlivněn farmakogenetickou výbavou, mnohdy má farmakogenetika i přímý dopad na prognózu nemocného.
Význam farmakogenetiky v inhibici sympatoadrenální aktivity betablokátory
Blokátory β-adrenergních receptorů jsou v léčbě srdečního selhání skupinou s nejlépe doloženým účinkem v řadě specifických situací – od asymptomatické insuficience levé komory až po nejvážněji nemocné.
Betablokátory je možno rozdělit na lipofilní a hydrofilní. Hydrofilní betablokátory, např. bisoprolol, betaxolol aj., jsou vylučovány ledvinami, aniž procházejí předchozí biotransformací. Lipofilní betablokátory, jako je metoprolol, karvedilol a nebivolol, jsou po biotransformaci izoenzymem CYP2D6 vylučovány játry do žluče. Lipofilní betablokátory jsou před eliminací přeměněny na hydrofilní metabolit oxidázou CYP2D6. Tato konverze však je problematická, ve velkém procentu populace se vyskytují polymorfismy měnící její aktivitu. Analýza české populace ukazuje, že pomalých metabolizátorů s žádnou funkční alelou (genotyp CYP2D6*3 či *4, event. *5) se vyskytuje asi 7 %, středních s jednou funkční alelou (genotyp CYP2D6*9 či *10, ev. *17) je 35–40 %, rychlých metabolizátorů s oběma funkčními alelami (genotyp CYP2D6*1) je 50 % a velmi rychlí metabolizátoři s multiplikací genu (genotyp CYP2D6*2) tvoří asi 6 % populace [7].
Důsledkem je, že pro 10–15 % populace nevyhovuje běžné dávkovací schéma léků – asi pro 5–10 % jsou dávky
![Graf 2 Vliv polymorfismu CYP2C9 na expozici (AUC) metoprololu; podle [8, 9] – Kirchheiner, et al., 2004, Seeringer, et al., 2008. Mezi skupinou rychlých a pomalých metabolizátorů existují více než desetinásobné interindividuální rozdíly v expozici. U pomalých metabolizátorů zůstává metoprolol v terapeutickém rozmezí 15–20 hodin, u ultrarychlých pouze 2–3 hodiny.](https://www.remedia.cz/photo-a-29258---.jpg) příliš vysoké, a naopak pro 5–10 % příliš nízké. Rozdíly v expozici (AUC) lipofilním betablokátorům se u obou variant liší 10–30násobně [8, 9]. Jinak řečeno, např. biologický poločas eliminace metoprololu se pohybuje u pomalých metabolizátorů kolem 10 hodin a u velmi rychlých kolem 2 hodin (graf 2). Rozdíl v maximální plazmatické koncentraci metoprololu mezi pomalými metabolizátory a běžnou variantou populace (střední či rychlí metabolizátoři) byl asi pětinásobný [10]. U velmi rychlých metabolizátorů je nutno počítat s tím, že při užití běžných dávek nedosáhneme terapeutického cíle, léčba u 5–10 % pacientů proto selhává. Variabilita expozice lipofilním betablokátorům je dána jak rozdílem v biologické dostupnosti, tak rozdílem v eliminaci. Při srovnání dopadu polymorfismu CYP2D6 na farmakokinetiku metoprololu, nebivololu a karvedilolu pozorujeme velké rozdíly – nejvíce je ovlivněna expozice metoprololu (více než desetinásobné rozdíly), nejméně pak expozice karvedilolu (maximálně šestinásobné rozdíly).
příliš vysoké, a naopak pro 5–10 % příliš nízké. Rozdíly v expozici (AUC) lipofilním betablokátorům se u obou variant liší 10–30násobně [8, 9]. Jinak řečeno, např. biologický poločas eliminace metoprololu se pohybuje u pomalých metabolizátorů kolem 10 hodin a u velmi rychlých kolem 2 hodin (graf 2). Rozdíl v maximální plazmatické koncentraci metoprololu mezi pomalými metabolizátory a běžnou variantou populace (střední či rychlí metabolizátoři) byl asi pětinásobný [10]. U velmi rychlých metabolizátorů je nutno počítat s tím, že při užití běžných dávek nedosáhneme terapeutického cíle, léčba u 5–10 % pacientů proto selhává. Variabilita expozice lipofilním betablokátorům je dána jak rozdílem v biologické dostupnosti, tak rozdílem v eliminaci. Při srovnání dopadu polymorfismu CYP2D6 na farmakokinetiku metoprololu, nebivololu a karvedilolu pozorujeme velké rozdíly – nejvíce je ovlivněna expozice metoprololu (více než desetinásobné rozdíly), nejméně pak expozice karvedilolu (maximálně šestinásobné rozdíly). Klinickým korelátem příliš nízké dávky betablokátoru je zpravidla nedostatečné snížení srdeční frekvence a markerem vysoké expozice je naopak bradykardie. U většiny nemocných můžeme dávku korigovat pouze změřením pulsové frekvence. Bohužel v praxi této výhody nevyužíváme a místo individuální úpravy volíme dávku doporučenou schválenými odbornými postupy. Reálná praxe ukazuje až stonásobné rozdíly v koncentraci metoprololu v léčené populaci, není respektována potřeba případné úpravy [11]. Obdobně v Rotterdamské studii nebyla pozorována korekce dávky podle srdeční frekvence, většina nemocných byla léčena „doporučenou“ dávkou bez ohledu na příliš nízkou či příliš vysokou srdeční frekvenci [12]. Analýzy kohortních studií ukazují, že výskyt převodních poruch u pomalých metabolizátorů je zvýšen asi pětinásobně. Jaký je dopad variability CYP2D6 u nemocných se srdečním selháním na akutní levostrannou insuficienci při iniciaci léčby, nevíme. Dá se předpokládat, že náhlý pokles kontraktility při rychlé expozici lipofilním betablokátorům u pomalých metabolizátorů může vést k neočekávanému zhoršení funkce levé komory. Postupná titrace dávky u pomalých metabolizátorů je tak zpochybněna, již iniciální dávka může vést k plné terapeutické koncentraci.
Nežádoucímu dopadu farmakogenetických rozdílů se vyhneme užitím hydrofilních betablokátorů (bisoprololu či betaxololu) s minimální interindividuální variabilitou efektu nebo zmírníme dopad užitím specifických lékových forem metoprololu s řízeným uvolňováním (ve formě ZOK).
Mluvíme-li o farmakogenetice v léčbě betablokátory, je nutno upozornit, že také odpověď na stimulaci či inhibici adrenergních receptorů je ovlivněna genetickou výbavou, tedy polymorfismy na úrovni adrenergního receptoru β a α. Tyto polymorfismy, kterých je popsáno více než šedesát, se v populaci objevují v desítkách procent a často ovlivňují tonus sympatického nervového systému i léčebnou odpověď na stimulaci či inhibici. Takovýto případ polymorfismu pre- a postsynaptického receptoru byl již uveden v obecné části. Jiný častý polymorfismus (≈ 15 % populace) receptoru β1 typu Ser49Gly zvyšuje down-regulaci receptoru po stimulaci (desenzibilizace), zabývali jsme se jím v obecné části.
Obdobných polymorfismů presynaptických a postsynaptických receptorů, které modulují afinitu noradrenalinu k inhibičnímu či aktivačnímu receptoru, je více. Některé z nich několikanásobně zvyšují citlivost ke stimulaci či k blokádě postsynaptických receptorů β1, β2, β3, α1 a α2. Jejich výskyt je významný, jsou přítomny asi u třetiny evropské populace a přispívají tak k rozdílům v terapeutickém účinku betablokátorů [13]. Do jaké míry se tyto polymorfismy uplatňují v etiopatogenezi či v modulaci průběhu kardiovaskulárních onemocnění, bude nutno teprve doložit.
Shrneme-li, pak farmakokinetika lipofilních betablokátorů (zejména metoprololu a nebivololu, méně již karvedilolu) je výrazně ovlivněna polymorfismem oxidázy CYP2D6. Klinický význam má přítomnost nízké či žádné aktivity u pomalých metabolizátorů (5–10 % populace). Zde je nutno počítat s významně vyšší expozicí, s prodloužením efektu a s vyšším výskytem nežádoucích účinků. Naopak u velmi rychlých metabolizátorů (5–8 % populace) je nutno počítat s nízkou expozicí lipofilním betablokátorům a se zkrácením účinku, farmakologický účinek bez adjustace dávky (např. podle srdeční frekvence) může selhat. Lpění na dodržení fixní cílové dávky lipofilních betablokátorů tak, jak je uvádějí doporučené postupy, nemusí být zcela racionální. Přehled o účinku beta-blokátoru u daného nemocného získáme např. sledováním srdeční frekvence. Její hodnota může vést k úpravě dávky.
Význam farmakogenetiky v inhibici RAAS
V inhibici RAAS v indikaci srdečního selhání užíváme tři skupiny léků – inhibitory ACE, blokátory receptorů AT1 (sartany) a blokátory aldosteronových receptorů spironolakton a eplerenon. Vzhledem k relativně nekomplikovanému metabolismu léků ovlivňujících RAAS se nesetkáváme s významným dopadem farmakogenetických odlišností na farmakokinetiku.
Genetická výbava však moduluje aktivitu RAAS i odpověď na léčbu. Významné jsou změny v aktivitě systému na úrovni angiotenzinogenu, ACE, na úrovni receptoru AT1, receptoru MAS či receptorů kininových. Polymorfismy na této úrovni vedou k silnější nebo naopak slabší odpovědi na stimulaci nebo snižují afinitu léků k cílovým strukturám [14].
Pozorování zhoršené prognózy nemocných se srdečním selháním u varianty ACE typu Del (rs1799752) s vysokou aktivitou RAAS bude nutno ověřit. Rovněž zde však, a to i v případě potvrzení nálezu, bude klinický dopad malý, léčba betablokátory či inhibitory ACE rozdíl mezi variantami Del/Ins zcela smazala. Jinak řečeno, inhibice hyperaktivovaného RAAS vedla, v souladu s očekáváním, k úpravě prognózy. Přínos z léčby nemocných s touto variantou ACE byl významně větší.
Inhibitory ACE
Mechanismus účinku inhibitorů ACE je pleiotropní. Vedle snížení nabídky angiotenzinu II s nižší stimulací receptoru AT1 se uplatní jak zpomalení biodegradace bradykininu se zvýšením aktivace bradykininových receptorů BK1 a BK2, tak zvýšení nabídky angiotenzinu 1–7 stimulující receptory MAS. Jak receptory bradykininové, tak receptory MAS navodí vazodilataci a natriurézu, výsledně tak oslabují efekt stimulace receptoru AT1.
Při modulaci efektu inhibitorů ACE se také významně uplatní genotyp. Máme k dispozici recentní analýzu účinku perindoprilu na prognózu v závislosti na farmakogenetické výbavě u nemocných s ischemickou chorobou srdeční z podstudie PERGENE. Tato farmakogenetická analýza byla provedena u více než 9 tisíc nemocných sledovaných v rámci studie EVROPA [15]. Na základě sledování efektu 12 polymorfních kandidátních genů, které by mohly ovlivňovat prognózu léčených nemocných, byly vytipovány nejméně tři polymorfismy ovlivňující dopad léčby. Významné byly zejména polymorfismy měnící aktivitu receptoru AT1 (varianty rs275651 a rs5182) a receptoru BK1 (rs12050217). Nemocní s kardiovaskulárním postižením a s přítomností méně rizikových alel, maximálně dvou (tři čtvrtiny populace), měli z léčby inhibitorem ACE prospěch, nosiči více rizikových alel nikoli (obr. 3). Potvrdí-li se pozorování studie, pak se naskýtá cesta pro individualizaci léčby inhibitory ACE na základě farmakogenetického vyšetření.
Metaanalýza 19 studií sledujících farmakogenetickou asociaci nežádoucích účinků inhibitorů ACE dokládá vazbu některých polymorfismů s výskytem angioedému, nikoli však kašle [16]. Klinický význam je nepravděpodobný.
Sartany
Při léčbě sartany se setkáváme s ovlivněním farmakokinetiky farmakogenetickou výbavou. V oblasti bioaktivace je významná interakce a závislost u losartanu. Losartan je proléčivo, 10–20 % je konvertováno na asi 30krát aktivnější metabolit E 3174, většina farmakodynamického účinku je tak zprostředkována tímto metabolitem s významně delším biologickým poločasem eliminace. Konverze je zprostředkována zejména izoenzymem CYP2C9, který je významně polymorfní. Mutace CYP2C9*2 a CYP2C9*3, které se objevují asi u 30 % indoevropské populace v heterozygotní podobě a přibližně u 2 % v homozygotní formě, mají nižší aktivitu ve srovnání s běžným genotypem CYP2C9*1. Genotypy s nižší aktivitou CYP2C9 jsou spojeny se sníženou bioaktivací losartanu, např. u nositelů CYP2C9*3/*3 je výsledná koncentrace aktivního metabolitu zhruba 10 % [17, 18]. Z tohoto důvodu je k zajištění spolehlivějšího účinku vhodná volba jiných sartanů, například kandesartanu.
Blokátory mineralokortikoidních receptorů
Na úrovni inhibice aldosteronových receptorů spironolaktonem či eplerenonem není zjištěn vliv farmakogenetické výbavy na účinek léčby.
Význam farmakogenetiky při léčbě diuretiky
Vliv genetické výbavy se projeví v oblasti diuretik jen minimálně. Pouze furosemid je substrátem P-gp, vyšší, či naopak nízká aktivita této pumpy významně ovlivní výslednou biologickou dostupnost, jednou se pohybuje kolem 10 %, jindy se blíží 100 % [19]. Naštěstí přímý klinický význam není velký. Furosemid má tak velké terapeutické okno, že se zvýšení expozice projeví jen mírnou akcentací diurézy, toxický efekt nebývá přítomen. Přímým důsledkem je pouze nutnost iniciální titrace dávky.
Význam farmakogenetiky u léčiv s pozitivně inotropním účinkem
Inotropika můžeme – podle mechanismu účinku – rozdělit do dvou skupin: inotropika stimulující kontraktilitu zvýšením sarkoplazmatické koncentrace kalcia (typicky digitalisové alkaloidy – digoxin, či β1-sympatomimetika – adrenalin, dopamin aj.) nebo inotropika působící zvýšením afinity kalcia k regulačnímu proteinu – troponinu C – čili senzibilizátory kalcia (levosimendan aj.). Prvá skupina, zejména digoxin, má velmi úzké terapeutické okno – již malý vzestup koncentrace digoxinu zvýší intracelulární hladinu kalcia a významně stimuluje excitabilitu myokardu s rizikem vzniku maligních arytmií. Podobně je s rizikem maligních tachyarytmií spojena léčba katecholaminy.
Digoxin
Digoxin je typickým lékem s úzkým terapeutickým oknem, kde již mírné poddávkování vede k selhání efektu, a naopak předávkování je spojeno s toxickým účinkem a zvýšením mortality. Post-hoc analýza studie DIG, která sledovala vztah plazmatických koncentrací digoxinu k mortalitě, ukázala, že koncentrace 0,5–0,8 ng/ml
byla spojena se signifikantním snížením mortality, koncentrace pohybující se v rozmezí 0,9–1,1 ng/ml, stejně jako koncentrace nižší než 0,5 ng/ml, mortalitu neovlivnily a zvýšení hladiny nad 1,2 ng/ml bylo spojeno se vzestupem mortality ve srovnání s placebem [20]. Ke stejnému nálezu, tj. zjištění, že ovlivnění mortality digoxinem je závislé na jeho plazmatické koncentraci, dospěli i jiní autoři [21]. Absence kontroly hladiny digoxinu (recentně např. u nemocných s fibrilací síní s rychlou odpovědí komor) zvyšuje mortalitu významně [22]. Optimální terapeutické rozmezí digoxinu je velmi úzké, zpravidla se držíme hodnot 0,5–0,8 ng/ml, respektive podle doporučení Evropské kardiologické společnosti (ESC) volnějšího rozmezí 0,6–1,2 ng/ml.
Hladina digoxinu je ovlivněna zejména variabilní biologickou dostupností a variabilní renální eliminací (převážně tubulární sekrecí, méně glomerulární filtrací). Rozhodujícím faktorem absorpce i eliminace je aktivita P-gp, digoxin není v organismu významně transformován. Genotypy s nízkou aktivitou P-gp jsou spojeny s mírně vyšší dostupností digoxinu a naopak. Při analýze studií sledujících klinický význam farmakogenetické variability aktivity P-gp je patrno, že variabilita aktivity je jen méně významná, zvýšení expozice a plazmatické koncentrace nepřesahovalo 50 %. Naopak inhibice P-gp při lékových interakcích se uplatní výrazně více a klinický dopad je jednoznačně významný. Silné či střední inhibitory P-gp, konkrétně verapamil, amiodaron, dronedaron, propafenon, telmisartan, cyklosporin, azolová antimykotika (zejména itrakonazol a ketokonazol), a makrolidová antibiotika (zejména klarithromycin) zvyšují koncentraci digoxinu o 50 % až 150 %.
Sympatomimetika
Parenterálně aplikovaná sympatomimetika – adrenalin, noradrenalin, dopamin, dobutamin, isoprenalin – působí aktivací jednoho či více adrenergních receptorů α, β nebo receptorů dopaminových. Společným biodegradačním krokem těchto sympatomimetik je inaktivace monoaminooxidázou (MAO), přítomnou v řadě tkání, či katechol-O-methyltransferázou (COMT) v játrech. Význam farmakogenetiky v léčbě sympatomimetiky není doložen, naopak inhibice těchto enzymů (zejména antidepresivy) se uplatní významně.
Senzibilizátory kalcia
Farmakodynamický účinek levosimendanu není směřován ke zvýšení sarkoplazmatické hladiny kalcia, je zprostředkován stabilizací komplexu kalcium-troponin C s následným prodloužením aktino-myosinového spojení. Význam farmakogenetiky není u levosimendanu doložen. K bioaktivaci na aktivní metabolit je sice zapojena polymorfní N‑acetyltransferáza, rozdíly v její aktivitě se však klinicky uplatní jen minimálně, neboť farmakologický efekt má jak mateřská látka, tak aktivní metabolity.
Závěr
Podíváme-li se na uvedená fakta, pak význam farmakogenetiky pro průběh srdečního selhání a farmakodynamickou odpověď jednoznačně doložen není, vlivů je zřejmě mnoho a zatím známe jen málo dílků výsledné skládačky. Naopak ovlivnění farmakokinetiky řady léčiv genetickými faktory je zřetelné. Minimálně to u srdečního selhání platí v léčbě lipofilními betablokátory. Když se však zaměříme na léčiva určená nikoli specificky k terapii srdečního selhání, ale k léčbě dysrytmií, dyslipidemií či k potlačení trombotických a tromboembolických komplikací, uplatní se polymorfismy v absorpci a biotransformaci léčiv obdobně.
Riziko výběru léků s významným vlivem farmakogenetiky snížíme dodržováním několika následujících zásad. Při volbě léků bychom měli upřednostnit léky s vyšší biologickou dostupností. Zvýšení dostupnosti při inhibici transportních a metabolických systémů se pak projeví jen menším vzestupem koncentrace. Příkladem je preference atorvastatinu s dostupností 15–20 % proti simvastatinu s 3–5% dostupností. Zvýšení biologické dostupnosti při hyperaktivitě nebo inhibici P-gp i izoenzymu CYP3A4 zvýší koncentraci atorvastatinu 3–4násobně, zatímco koncentraci simvastatinu 10–20násobně.
Další možností je volba léků s alternativními a nekomplikovanými cestami bioaktivace a biodegradace. Příkladem je volba prasugrelu nebo tikagreloru místo klopidogrelu, bisoprololu místo metoprololu, respektive dabigatranu či rivaroxabanu místo warfarinu.
Konečně v indikovaných případech je možno volit přímé stanovení polymorfismu metabolických systémů. Tento přístup je zatím běžný při použití některých cytostatik a imunosupresiv, v kardiologii však farmakogenetické testování běžné není. Tam, kde existují problémy, například při podávání warfarinu (testování oxidázy CYP2C9) nebo klopidogrelu (P-gp a CYP2C19), je situace natolik komplikovaná a do výsledného efektu zasahuje tolik faktorů, že stanovení genotypu, resp. fenotypu, neposkytuje zásadní přínos v rozhodovacím procesu.
Seznam použité literatury
- [1] Voora D, Shah SH, Spasojevic I, et al. The SLCO1B1*5 genetic variant is associated with statin-induced side effects. J Am Coll Cardiol 2009; 54: 1609–1616.
- [2] Link E, Parish S, Armitage J, et al. SLCO1B1 variants and statin-induced myopathy – a genome-wide study. N Engl J Med 2008; 359: 789–799.
- [3] Sakaeda T, Nakamura T, Okumura K. MDR1 genotype-related pharmacokinetics and pharmacodynamics. Biol Pharm Bull 2002; 25: 1391–1400.
- [4] Parry HM, Doney AS, Palmer CN, Lang CC. State of play of pharmacogenetics and personalized medicine in heart failure. Cardiovasc Ther 2013 Mar 11. Epub ahead of print.
- [5] Mann DL, Kent RL, Parsons B, et al Adrenergic effects on the biology of the adult mammalian cardiocyte. Circulation 1992; 85: 790e804.
- [6] Levin MC, Marullo S, Muntaner O, et al. The myocardium-protective Gly-49 variant of the beta 1-adrenergic receptor exhibits constitutive activity and increased desensitization and down-regulation. J Biol Chem 2002; 277: 30429e35.
- [7] Buzkova H, et al. Frequency of single nucleotide polymorphisms of CYP2D6 in the Czech population. Cell Biochemistry Function 2008; 26: 76–81.
- [8] Kirchheiner J, et al. Impact of the ultrarapid metaboliser genotype of cytochrome P450 2D6 on metoprolol pharmacokinetics and pharmacodynamics. Clin Pharmacol Ther 2004; 76: 302–312.
- [9] Seeringer A, Brockmöller J, Bauer S, et al. Enantiospecific pharmacokinetics of metoprolol in CYP2D6 ultra-rapid metabolisers and correlation with exercise-induced heart rate. Eur J Clin Pharmacol 2008; 64: 883–888.
- [10] Rau T, Wuttke H, Michels LM, et al. Impact of the CYP2D6 genotype on the clinical effects of metoprolol: a prospective longitudinal study. Clin Pharmacol Ther 2009; 85: 269–272.
- [11] Ismail R, Teh LK. The relevance of CYP2D6 genetic polymorphism on chronic metoprolol therapy in cardiovascular patients. J Clin Pharm Ther 2006; 31: 99–109.
- [12] Bijl MJ, Visser LE, van Schaik RH. Genetic variation in the CYP2D6 gene is associated with a lower heart rate and blood pressure in beta-blocker users. Clin Pharmacol Ther 2009; 85: 45–50.
- [13] von Homeyer P, Schwinn DA. Review Articles: Pharmacogenomics of β-Adrenergic Receptor Physiology and Response to β-Blockade. Anesth Analg 2011; 113: 1305–1318.
- [14] Talameh JA, McLeod HL, Adams KF Jr, Patterson JH. Genetic tailoring of pharmacotherapy in heart failure: optimize the old, while we wait for something new. J Card Fail 2012; 18: 338–349.
- [15] Brugts JJ, de Maat MP, Danser AH, et al. Individualised therapy of angiotensin converting enzyme (ACE) inhibitors in stable coronary artery disease: overview of the primary results of the PERindopril GENEtic association (PERGENE) study. Neth Heart J 2012; 20: 24–32.
- [16] Mahmoudpour SH, Leusink M, Putten L, et al. Pharmacogenetics of ACE inhibitor-induced angioedema and cough: a systematic review and meta-analysis. Pharmacogenomics 2013; 14: 249–260.
- [17] Zhou SF, Zhou ZW, Huang M. Polymorphisms of human cytochrome P450 2C9 and the functional relevance. Toxicology 2010; 278: 165–188.
- [18] Wang B, Wang J, Huang SQ, et al. Genetic polymorphism of the human cytochrome P450 2C9 gene and its clinical significance. Curr Drug Metab 2009; 10: 781–834.
- [19] Murray MD, Haag KM, Black PK, et al. Variable furosemide absorption and poor predictability of response in elderly patients. Pharmacotherapy 1997; 17: 98–106.
- [20] Rathore SS, Curtis JP, Wang Y et al. Association of serum digoxin concentration and outcomes in patients with heart failure. JAMA 2003; 289: 871–878.
- [21] Ahmed A, Rich MW, Love TE, et al. Digoxin and reduction in mortality and hospitalization in heart failure: a comprehensive post hoc analysis of the DIG trial. Eur Heart J 2006; 27: 178–186.
- [22] Heartwire 2013: Another strike for digoxin in AF: Mortality goes up in cohort study, analýza studie AFFIRM a kohortní studie přednesené na kongresu ACC 2013.